Idiopathic pulmonary fibrosis (IPF) is a disease that causes scarring and fibrotic transformation of the lung parenchyma, resulting in the progressive loss of respiratory function and, often, death. An increasing body of literature shows that pulmonary vascular permeability may play a big role in the pathogenesis of this condition. There is a search for therapeutic targets to try and modulate this vascular permeability in fibrotic lungs. One such class of targets that shows great promise is sphingolipids. In this review, we examine the ways that sphingolipids can affect vascular permeability and the pathogenesis of IPF, as well as the pros and cons of their potential role as therapeutic targets for treating IPF.
- idiopathic pulmonary fibrosis
- sphingolipids
- sphingosine-1-phosphate
1. Introduction
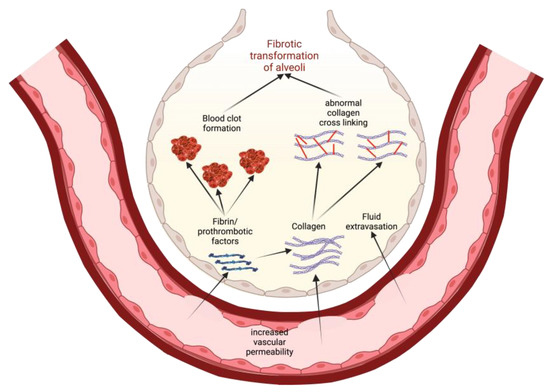
2. Models of Pathogenesis of Idiopathic Pulmonary Fibrosis
3. The Endothelial Barrier
4. The Lung Endothelium in Inflammatory States
5. Rho Kinases
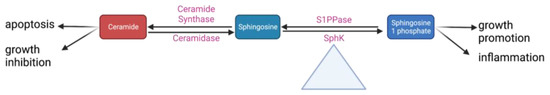
6. Sphingolipids
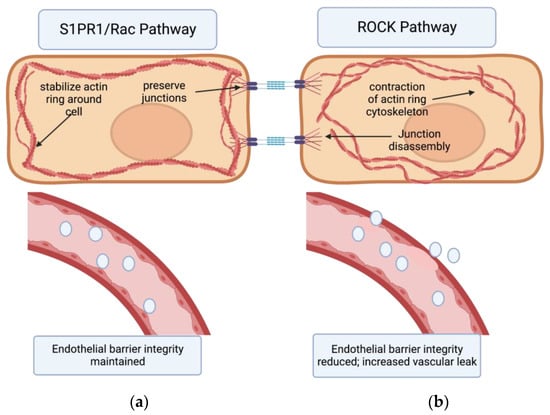
7. The Effects of S1PR1 on Vascular Permeability
8. The Effects of Other S1P Receptors and Sphingolipids on Vascular Permeability
9. S1P Modulators and Sphingolipid-Based Treatment Options
10. Angiogenesis in IPF
11. Future Study
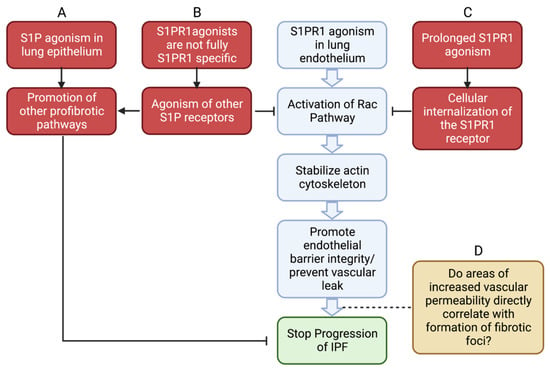
12. Conclusions
This entry is adapted from the peer-reviewed paper 10.3390/biomedicines11061728
References
- Maher, T.M.; Bendstrup, E.; Dron, L.; Langley, J.; Smith, G.; Khalid, J.M.; Patel, H.; Kreuter, M. Global incidence and prevalence of idiopathic pulmonary fibrosis. Respir. Res. 2021, 22, 197.
- Zheng, Q.; Cox, I.A.; Campbell, J.A.; Xia, Q.; Otahal, P.; de Graaff, B.; Corte, T.J.; Teoh, A.K.Y.; Walters, E.H.; Palmer, A.J. Mortality and survival in idiopathic pulmonary fibrosis: A systematic review and meta-analysis. ERJ Open Res. 2022, 8, 00591–2021.
- Nathan, S.D.; Albera, C.; Bradford, W.Z.; Costabel, U.; Glaspole, I.; Glassberg, M.K.; Kardatzke, D.R.; Daigl, M.; Kirchgaessler, K.-U.; Lancaster, L.H.; et al. Effect of pirfenidone on mortality: Pooled analyses and meta-analyses of clinical trials in idiopathic pulmonary fibrosis. Lancet Respir. Med. 2017, 5, 33–41.
- Spagnolo, P.; Kropski, J.A.; Jones, M.G.; Lee, J.S.; Rossi, G.; Karampitsakos, T.; Maher, T.M.; Tzouvelekis, A.; Ryerson, C.J. Idiopathic pulmonary fibrosis: Disease mechanisms and drug development. Pharmacol. Ther. 2021, 222, 107798.
- Noble, P.W.; Albera, C.; Bradford, W.Z.; Costabel, U.; Glassberg, M.K.; Kardatzke, D.; King, T.E.; Lancaster, L.; Sahn, S.A.; Szwarcberg, J.; et al. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): Two randomised trials. Lancet 2011, 377, 1760–1769.
- Taniguchi, H.; Ebina, M.; Kondoh, Y.; Ogura, T.; Azuma, A.; Suga, M.; Taguchi, Y.; Takahashi, H.; Nakata, K.; Sato, A.; et al. Pirfenidone in idiopathic pulmonary fibrosis. Eur. Respir. J. 2010, 35, 821–829.
- Canestaro, W.J.; Forrester, S.H.; Raghu, G.; Ho, L.; Devine, B.E. Drug Treatment of Idiopathic Pulmonary Fibrosis: Systematic Review and Network Meta-Analysis. Chest 2016, 149, 756–766.
- Ahluwalia, N.; Shea, B.S.; Tager, A.M. New therapeutic targets in idiopathic pulmonary fibrosis. Aiming to rein in runaway wound-healing responses. Am. J. Respir. Crit. Care Med. 2014, 190, 867–878.
- Yang, J.; Pan, X.; Wang, L.; Yu, G. Alveolar cells under mechanical stressed niche: Critical contributors to pulmonary fibrosis. Mol. Med. 2020, 26, 95.
- Hewlett, J.C.; Kropski, J.A.; Blackwell, T.S. Idiopathic pulmonary fibrosis: Epithelial-mesenchymal interactions and emerging therapeutic targets. Matrix Biol. 2018, 71–72, 112–127.
- McKeown, S.; Richter, A.G.; O’Kane, C.; McAuley, D.F.; Thickett, D.R. MMP expression and abnormal lung permeability are important determinants of outcome in IPF. Eur. Respir. J. 2009, 33, 77–84.
- Declercq, M.; Treps, L.; Carmeliet, P.; Witters, P. The role of endothelial cells in cystic fibrosis. J. Cyst. Fibros. 2019, 18, 752–761.
- Murakami, A.; Takasugi, H.; Ohnuma, S.; Koide, Y.; Sakurai, A.; Takeda, S.; Hasegawa, T.; Sasamori, J.; Konno, T.; Hayashi, K.; et al. Sphingosine 1-phosphate (S1P) regulates vascular contraction via S1P3 receptor: Investigation based on a new S1P3 receptor antagonist. Mol. Pharmacol. 2010, 77, 704–713.
- Huang, L.S.; Sudhadevi, T.; Fu, P.; Punathil-Kannan, P.-K.; Ebenezer, D.L.; Ramchandran, R.; Putherickal, V.; Cheresh, P.; Zhou, G.; Ha, A.W.; et al. Sphingosine Kinase 1/S1P Signaling Contributes to Pulmonary Fibrosis by Activating Hippo/YAP Pathway and Mitochondrial Reactive Oxygen Species in Lung Fibroblasts. Int. J. Mol. Sci. 2020, 21, 2064.
- Khan, S.A.; Goliwas, K.F.; Deshane, J.S. Sphingolipids in Lung Pathology in the Coronavirus Disease Era: A Review of Sphingolipid Involvement in the Pathogenesis of Lung Damage. Front. Physiol. 2021, 12, 760638.
- Jernigan, P.L.; Makley, A.T.; Hoehn, R.S.; Edwards, M.J.; Pritts, T.A. The role of sphingolipids in endothelial barrier function. Biol. Chem. 2015, 396, 681–691.
- Kuperberg, S.J.; Wadgaonkar, R. Sepsis-Associated Encephalopathy: The Blood-Brain Barrier and the Sphingolipid Rheostat. Front. Immunol. 2017, 8, 597.
- Knipe, R.S.; Spinney, J.J.; Abe, E.A.; Probst, C.K.; Franklin, A.; Logue, A.; Giacona, F.; Drummond, M.; Griffith, J.; Brazee, P.L.; et al. Endothelial-Specific Loss of Sphingosine-1-Phosphate Receptor 1 Increases Vascular Permeability and Exacerbates Bleomycin-induced Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2022, 66, 38–52.
- Knipe, R.S.; Spinney, J.J.; Abe, E.; Probst, C.K.; Logue, A.; Griffith, J.; Black, K.E.; Montesi, S.B.; Shea, B.; Medoff, B.D. Loss of endothelial S1PR1 exacerbates bleomycin-induced pulmonary fibrosis through intra-alveolar coagulation and immune cell infiltration. Am. J. Respir. Crit. Care Med. 2020, 201, A7878.
- Wadgaonkar, R.; Patel, V.; Grinkina, N.; Romano, C.; Liu, J.; Zhao, Y.; Sammani, S.; Garcia, J.G.N.; Natarajan, V. Differential regulation of sphingosine kinases 1 and 2 in lung injury. Am. J. Physiol. Lung Cell Mol. Physiol. 2009, 296, L603–L613.
- Hay, J.; Shahzeidi, S.; Laurent, G. Mechanisms of bleomycin-induced lung damage. Arch. Toxicol. 1991, 65, 81–94.
- Probst, C.K.; Montesi, S.B.; Medoff, B.D.; Shea, B.S.; Knipe, R.S. Vascular permeability in the fibrotic lung. Eur. Respir. J. 2020, 56, 1900100.
- Villar, J.; Zhang, H.; Slutsky, A.S. Lung Repair and Regeneration in ARDS: Role of PECAM1 and Wnt Signaling. Chest 2019, 155, 587–594.
- Engelbrecht, E.; Kooistra, T.; Knipe, R.S. The Vasculature in Pulmonary Fibrosis. Curr. Tissue Microenviron. Rep. 2022, 3, 83–97.
- Komarova, Y.; Malik, A.B. Regulation of endothelial permeability via paracellular and transcellular transport pathways. Annu. Rev. Physiol. 2010, 72, 463–493.
- Pober, J.S.; Sessa, W.C. Evolving functions of endothelial cells in inflammation. Nat. Rev. Immunol. 2007, 7, 803–815.
- Mehta, D.; Malik, A.B. Signaling mechanisms regulating endothelial permeability. Physiol. Rev. 2006, 86, 279–367.
- Campbell, H.K.; Maiers, J.L.; DeMali, K.A. Interplay between tight junctions & adherens junctions. Exp. Cell. Res. 2017, 358, 39–44.
- De Caterina, R.; Libby, P.; Peng, H.B.; Thannickal, V.J.; Rajavashisth, T.B.; Gimbrone, M.A.; Shin, W.S.; Liao, J.K. Nitric oxide decreases cytokine-induced endothelial activation. Nitric oxide selectively reduces endothelial expression of adhesion molecules and proinflammatory cytokines. J. Clin. Investig. 1995, 96, 60–68.
- Claesson-Welsh, L. Vascular permeability—The essentials. Ups. J. Med. Sci. 2015, 120, 135–143.
- Nagy, J.A.; Benjamin, L.; Zeng, H.; Dvorak, A.M.; Dvorak, H.F. Vascular permeability, vascular hyperpermeability and angiogenesis. Angiogenesis 2008, 11, 109–119.
- Wettschureck, N.; Strilic, B.; Offermanns, S. Passing the Vascular Barrier: Endothelial Signaling Processes Controlling Extravasation. Physiol. Rev. 2019, 99, 1467–1525.
- Jambusaria, A.; Hong, Z.; Zhang, L.; Srivastava, S.; Jana, A.; Toth, P.T.; Dai, Y.; Malik, A.B.; Rehman, J. Endothelial heterogeneity across distinct vascular beds during homeostasis and inflammation. eLife 2020, 9, e51413.
- Shimoda, L.A.; Semenza, G.L. HIF and the lung: Role of hypoxia-inducible factors in pulmonary development and disease. Am. J. Respir. Crit. Care Med. 2011, 183, 152–156.
- Laddha, A.P.; Kulkarni, Y.A. VEGF and FGF-2: Promising targets for the treatment of respiratory disorders. Respir. Med. 2019, 156, 33–46.
- Ramakrishnan, S.; Anand, V.; Roy, S. Vascular endothelial growth factor signaling in hypoxia and inflammation. J. Neuroimmune Pharmacol. 2014, 9, 142–160.
- Broermann, A.; Winderlich, M.; Block, H.; Frye, M.; Rossaint, J.; Zarbock, A.; Cagna, G.; Linnepe, R.; Schulte, D.; Nottebaum, A.F.; et al. Dissociation of VE-PTP from VE-cadherin is required for leukocyte extravasation and for VEGF-induced vascular permeability in vivo. J. Exp. Med. 2011, 208, 2393–2401.
- Nottebaum, A.F.; Cagna, G.; Winderlich, M.; Gamp, A.C.; Linnepe, R.; Polaschegg, C.; Filippova, K.; Lyck, R.; Engelhardt, B.; Kamenyeva, O.; et al. VE-PTP maintains the endothelial barrier via plakoglobin and becomes dissociated from VE-cadherin by leukocytes and by VEGF. J. Exp. Med. 2008, 205, 2929–2945.
- Oldenburg, J.; de Rooij, J. Mechanical control of the endothelial barrier. Cell Tissue Res. 2014, 355, 545–555.
- Nagatoya, K.; Moriyama, T.; Kawada, N.; Takeji, M.; Oseto, S.; Murozono, T.; Ando, A.; Imai, E.; Hori, M. Y-27632 prevents tubulointerstitial fibrosis in mouse kidneys with unilateral ureteral obstruction. Kidney Int. 2002, 61, 1684–1695.
- Knipe, R.S.; Probst, C.K.; Lagares, D.; Franklin, A.; Spinney, J.J.; Brazee, P.L.; Grasberger, P.; Zhang, L.; Black, K.E.; Sakai, N.; et al. The Rho Kinase Isoforms ROCK1 and ROCK2 Each Contribute to the Development of Experimental Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2018, 58, 471–481.
- Kitamura, K.; Tada, S.; Nakamoto, N.; Toda, K.; Horikawa, H.; Kurita, S.; Tsunematsu, S.; Kumagai, N.; Ishii, H.; Saito, H.; et al. Rho/Rho kinase is a key enzyme system involved in the angiotensin II signaling pathway of liver fibrosis and steatosis. J. Gastroenterol. Hepatol. 2007, 22, 2022–2033.
- Katoh, K.; Kano, Y.; Noda, Y. Rho-associated kinase-dependent contraction of stress fibres and the organization of focal adhesions. J. R. Soc. Interface 2011, 8, 305–311.
- Wojciak-Stothard, B.; Ridley, A.J. Rho GTPases and the regulation of endothelial permeability. Vascul. Pharmacol. 2002, 39, 187–199.
- Mong, P.Y.; Wang, Q. Activation of Rho kinase isoforms in lung endothelial cells during inflammation. J. Immunol. 2009, 182, 2385–2394.
- Abbès, M.; Sabatier, P. . Ann. Chir. Plast. 1970, 15, 205–213.
- Del Gaudio, I.; Camerer, E. Distinct GEFs Couple S1PR1 to Rac for Endothelial Barrier Enhancement and Lymphocyte Trafficking. Arterioscler Thromb. Vasc. Biol. 2022, 42, 903–905.
- Wadgaonkar, R.; Geraghty, P.; Kabir, I.; Foronjy, R. Role of sphingomyelin synthase regulated micro domain signaling in cigarette smoke induced inflammation. Am. J. Respir. Crit. Care Med. 2017, 195, A6339.
- Gowda, S.; Yeang, C.; Wadgaonkar, S.; Anjum, F.; Grinkina, N.; Cutaia, M.; Jiang, X.-C.; Wadgaonkar, R. Sphingomyelin synthase 2 (SMS2) deficiency attenuates LPS-induced lung injury. Am. J. Physiol. Lung Cell Mol. Physiol. 2011, 300, L430–L440.
- Wattenberg, B.W.; Pitson, S.M.; Raben, D.M. The sphingosine and diacylglycerol kinase superfamily of signaling kinases: Localization as a key to signaling function. J. Lipid Res. 2006, 47, 1128–1139.
- Siow, D.L.; Anderson, C.D.; Berdyshev, E.V.; Skobeleva, A.; Natarajan, V.; Pitson, S.M.; Wattenberg, B.W. Sphingosine kinase localization in the control of sphingolipid metabolism. Adv. Enzyme Regul. 2011, 51, 229–244.
- Bravo, G.A.; Cedeno, R.R.; Casadevall, M.P.; Ramio-Torrenta, L. Sphingosine-1-Phosphate (S1P) and S1P Signaling Pathway Modulators, from Current Insights to Future Perspectives. Cells 2022, 11, 2058.
- Sanchez, T.; Skoura, A.; Wu, M.T.; Casserly, B.; Harrington, E.O.; Hla, T. Induction of vascular permeability by the sphingosine-1-phosphate receptor-2 (S1P2R) and its downstream effectors ROCK and PTEN. Arterioscler Thromb. Vasc. Biol. 2007, 27, 1312–1318.
- Sanchez, T.; Hla, T. Structural and functional characteristics of S1P receptors. J. Cell. Biochem. 2004, 92, 913–922.
- Forrest, M.; Sun, S.Y.; Hajdu, R.; Bergstrom, J.; Card, D.; Doherty, G.; Hale, J.; Keohane, C.; Meyers, C.; Milligan, J.; et al. Immune cell regulation and cardiovascular effects of sphingosine 1-phosphate receptor agonists in rodents are mediated via distinct receptor subtypes. J. Pharmacol. Exp. Ther. 2004, 309, 758–768.
- Jin, F.; Hagemann, N.; Sun, L.; Wu, J.; Doeppner, T.R.; Dai, Y.; Hermann, D.M. High-density lipoprotein (HDL) promotes angiogenesis via S1P3-dependent VEGFR2 activation. Angiogenesis 2018, 21, 381–394.
- Gräler, M.H.; Grosse, R.; Kusch, A.; Kremmer, E.; Gudermann, T.; Lipp, M. The sphingosine 1-phosphate receptor S1P4 regulates cell shape and motility via coupling to Gi and G12/13. J. Cell. Biochem. 2003, 89, 507–519.
- Niedernberg, A.; Scherer, C.R.; Busch, A.E.; Kostenis, E. Comparative analysis of human and rat S1P(5) (edg8): Differential expression profiles and sensitivities to antagonists. Biochem. Pharmacol. 2002, 64, 1243–1250.
- Garcia, J.G.; Liu, F.; Verin, A.D.; Birukova, A.; Dechert, M.A.; Gerthoffer, W.T.; Bamberg, J.R.; English, D. Sphingosine 1-phosphate promotes endothelial cell barrier integrity by Edg-dependent cytoskeletal rearrangement. J. Clin. Investig. 2001, 108, 689–701.
- Berdyshev, E.V.; Gorshkova, I.; Usatyuk, P.; Kalari, S.; Zhao, Y.; Pyne, N.J.; Pyne, S.; Sabbadini, R.A.; Garcia, J.G.N.; Natarajan, V. Intracellular S1P generation is essential for S1P-induced motility of human lung endothelial cells: Role of sphingosine kinase 1 and S1P lyase. PLoS ONE 2011, 6, e16571.
- Adyshev, D.M.; Moldobaeva, N.K.; Elangovan, V.R.; Garcia, J.G.N.; Dudek, S.M. Differential involvement of ezrin/radixin/moesin proteins in sphingosine 1-phosphate-induced human pulmonary endothelial cell barrier enhancement. Cell Signal. 2011, 23, 2086–2096.
- Donati, C.; Bruni, P. Sphingosine 1-phosphate regulates cytoskeleton dynamics: Implications in its biological response. Biochim. Biophys. Acta 2006, 1758, 2037–2048.
- Sun, X.; Shikata, Y.; Wang, L.; Ohmori, K.; Watanabe, N.; Wada, J.; Shikata, K.; Birukov, K.G.; Makino, H.; Jacobson, J.R.; et al. Enhanced interaction between focal adhesion and adherens junction proteins: Involvement in sphingosine 1-phosphate-induced endothelial barrier enhancement. Microvasc. Res. 2009, 77, 304–313.
- Hla, T.; Brinkmann, V. Sphingosine 1-phosphate (S1P): Physiology and the effects of S1P receptor modulation. Neurology 2011, 76, S3–S8.
- Bazzoni, G.; Dejana, E. Endothelial cell-to-cell junctions: Molecular organization and role in vascular homeostasis. Physiol. Rev. 2004, 84, 869–901.
- Natarajan, V.; Dudek, S.M.; Jacobson, J.R.; Moreno-Vinasco, L.; Huang, L.S.; Abassi, T.; Mathew, B.; Zhao, Y.; Wang, L.; Bittman, R.; et al. Sphingosine-1-phosphate, FTY720, and sphingosine-1-phosphate receptors in the pathobiology of acute lung injury. Am. J. Respir. Cell Mol. Biol. 2013, 49, 6–17.
- Peng, X.; Hassoun, P.M.; Sammani, S.; McVerry, B.J.; Burne, M.J.; Rabb, H.; Pearse, D.; Tuder, R.M.; Garcia, J.G.N. Protective effects of sphingosine 1-phosphate in murine endotoxin-induced inflammatory lung injury. Am. J. Respir. Crit. Care Med. 2004, 169, 1245–1251.
- McVerry, B.J.; Peng, X.; Hassoun, P.M.; Sammani, S.; Simon, B.A.; Garcia, J.G.N. Sphingosine 1-phosphate reduces vascular leak in murine and canine models of acute lung injury. Am. J. Respir. Crit. Care Med. 2004, 170, 987–993.
- Knipe, R.S.; Spinney, J.J.; Abe, E.; Probst, C.K.; Franklin, A.; Griffith, J.W.; Liao, J.K.; McCarthy, J.R.; Shea, B.S.; Medoff, B.D. The pulmonary endothelium plays a critical role in the fibrotic response to lung injury through S1PR1 and rock mediated cytoskeletal rearrangements. Am. J. Respir. Crit. Care Med. 2019, 199, A4020.
- Shea, B.S.; Probst, C.K.; Brazee, P.L.; Rotile, N.J.; Blasi, F.; Weinreb, P.H.; Black, K.E.; Sosnovik, D.E.; Van Cott, E.M.; Violette, S.M.; et al. Uncoupling of the profibrotic and hemostatic effects of thrombin in lung fibrosis. JCI Insight 2017, 2, e86608.
- Milara, J.; Navarro, R.; Juan, G.; Peiro, T.; Serrano, A.; Ramon, M.; Morcillo, E.; Cortijo, J. Sphingosine-1-phosphate is increased in patients with idiopathic pulmonary fibrosis and mediates epithelial to mesenchymal transition. Thorax 2012, 67, 147–156.
- Brinkmann, V. FTY720 (fingolimod) in Multiple Sclerosis: Therapeutic effects in the immune and the central nervous system. Br. J. Pharmacol. 2009, 158, 1173–1182.
- Garnier, O.; Vilgrain, I. Dialogue between VE-Cadherin and Sphingosine 1 Phosphate Receptor1 (S1PR1) for Protecting Endothelial Functions. Int. J. Mol. Sci. 2023, 24, 4018.
- Leonard, A.; Grose, V.; Paton, A.W.; Paton, J.C.; Yule, D.I.; Rahman, A.; Fazal, F. Selective Inactivation of Intracellular BiP/GRP78 Attenuates Endothelial Inflammation and Permeability in Acute Lung Injury. Sci. Rep. 2019, 9, 2096.
- Shea, B.S.; Brooks, S.F.; Fontaine, B.A.; Chun, J.; Luster, A.D.; Tager, A.M. Prolonged exposure to sphingosine 1-phosphate receptor-1 agonists exacerbates vascular leak, fibrosis, and mortality after lung injury. Am. J. Respir. Cell Mol. Biol. 2010, 43, 662–673.
- Zhao, J.; Okamoto, Y.; Asano, Y.; Ishimaru, K.; Aki, S.; Yoshioka, K.; Takuwa, N.; Wada, T.; Inagaki, Y.; Takahashi, C.; et al. Sphingosine-1-phosphate receptor-2 facilitates pulmonary fibrosis through potentiating IL-13 pathway in macrophages. PLoS ONE 2018, 13, e0197604.
- Sammani, S.; Moreno-Vinasco, L.; Mirzapoiazova, T.; Singleton, P.A.; Chiang, E.T.; Evenoski, C.L.; Wang, T.; Mathew, B.; Husain, A.; Moitra, J.; et al. Differential effects of sphingosine 1-phosphate receptors on airway and vascular barrier function in the murine lung. Am. J. Respir. Cell Mol. Biol. 2010, 43, 394–402.
- Wang, Y.; Gao, T.-T.; Xu, D.-F.; Zhu, X.-Y.; Dong, W.-W.; Lv, Z.; Liu, Y.-J.; Jiang, L. Upregulation of sphingosine kinase 1 contributes to ventilator-associated lung injury in a two-hit model. Int. J. Mol. Med. 2019, 44, 2077–2090.
- Calabresi, P.A.; Radue, E.-W.; Goodin, D.; Jeffery, D.; Rammohan, K.W.; Reder, A.T.; Vollmer, T.; Agius, M.A.; Kappos, L.; Stites, T.; et al. Safety and efficacy of fingolimod in patients with relapsing-remitting multiple sclerosis (FREEDOMS II): A double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Neurol. 2014, 13, 545–556.
- Cohen, J.A.; Khatri, B.; Barkhof, F.; Comi, G.; Hartung, H.-P.; Montalban, X.; Pelletier, J.; Stites, T.; Ritter, S.; von Rosenstiel, P.; et al. Long-term (up to 4.5 years) treatment with fingolimod in multiple sclerosis: Results from the extension of the randomised TRANSFORMS study. J. Neurol. Neurosurg. Psychiatry 2016, 87, 468–475.
- Khatri, B.; Barkhof, F.; Comi, G.; Hartung, H.-P.; Kappos, L.; Montalban, X.; Pelletier, J.; Stites, T.; Wu, S.; Holdbrook, F.; et al. Comparison of fingolimod with interferon beta-1a in relapsing-remitting multiple sclerosis: A randomised extension of the TRANSFORMS study. Lancet Neurol. 2011, 10, 520–529.
- Lublin, F.; Miller, D.H.; Freedman, M.S.; Cree, B.A.C.; Wolinsky, J.S.; Weiner, H.; Lubetzki, C.; Hartung, H.-P.; Montalban, X.; Uitdehaag, B.M.J.; et al. Oral fingolimod in primary progressive multiple sclerosis (INFORMS): A phase 3, randomised, double-blind, placebo-controlled trial. Lancet 2016, 387, 1075–1084.
- Kong, Y.; Wang, H.; Wang, S.; Tang, N. FTY720, a sphingosine-1 phosphate receptor modulator, improves liver fibrosis in a mouse model by impairing the motility of bone marrow-derived mesenchymal stem cells. Inflammation 2014, 37, 1326–1336.
- Ni, H.; Chen, J.; Pan, M.; Zhang, M.; Zhang, J.; Chen, P.; Liu, B. FTY720 prevents progression of renal fibrosis by inhibiting renal microvasculature endothelial dysfunction in a rat model of chronic kidney disease. J. Mol. Histol. 2013, 44, 693–703.
- Qian, J.; Ye, Y.; Lv, L.; Zhu, C.; Ye, S. FTY720 attenuates paraquat-induced lung injury in mice. Int. Immunopharmacol. 2014, 21, 426–431.
- Liu, W.D.; Gao, G.; Liu, H.Y.; Yan, G.H.; Li, L.C.; Zhang, J.Y.; Cui, H. Effects of fty-720 on pulmonary fibrosis in mice via tgf-pl/p38 mapk/nf-kb signaling pathway. Chin. Pharmacol. Bull. 2020, 36, 250–256.
- Muller, H.C.; Hocke, A.C.; Hellwig, K.; Gutbier, B.; Peters, H.; Schonrock, S.M.; Tschernig, T.; Schmiedl, A.; Hippenstiel, S.; N’Guessan, P.D.; et al. The Sphingosine-1 Phosphate receptor agonist FTY720 dose dependently affected endothelial integrity in vitro and aggravated ventilator-induced lung injury in mice. Pulm. Pharmacol. Ther. 2011, 24, 377–385.
- Gendron, D.R.; Lemay, A.-M.; Lecours, P.B.; Perreault-Vallières, V.; Huppé, C.-A.; Bossé, Y.; Blanchet, M.-R.; Dion, G.; Marsolais, D. FTY720 promotes pulmonary fibrosis when administered during the remodelling phase following a bleomycin-induced lung injury. Pulm. Pharmacol. Ther. 2017, 44, 50–56.
- Sobel, K.; Menyhart, K.; Killer, N.; Renault, B.; Bauer, Y.; Studer, R.; Steiner, B.; Bolli, M.H.; Nayler, O.; Gatfield, J. Sphingosine 1-phosphate (S1P) receptor agonists mediate pro-fibrotic responses in normal human lung fibroblasts via S1P2 and S1P3 receptors and Smad-independent signaling. J. Biol. Chem. 2013, 288, 14839–14851.
- Keller, C.D.; Rivera Gil, P.; Tolle, M.; van der Giet, M.; Chun, J.; Radeke, H.H.; Schafer-Korting, M.; Kleuser, B. Immunomodulator FTY720 induces myofibroblast differentiation via the lysophospholipid receptor S1P3 and Smad3 signaling. Am. J. Pathol. 2007, 170, 281–292.
- Clemons, B.; Bain, G.; Lai, A.; Santini, A.M.; Goulet, L.; Boyett, M.; Roberts, E.; Rosen, H.; Opiteck, G.J.; Scott, F.L.; et al. Favourable S1P1R/5R selectivity profile of ozanimod confers safety benefit relating to S1P3R-mediated pro-fibrotic changes in fibroblasts. Mult. Scler. J. 2018, 24, 39.
- Pérez-Jeldres, T.; Alvarez-Lobos, M.; Rivera-Nieves, J. Targeting Sphingosine-1-Phosphate Signaling in Immune-Mediated Diseases: Beyond Multiple Sclerosis. Drugs 2021, 81, 985–1002.
- Karnati, S.; Seimetz, M.; Kleefeldt, F.; Sonawane, A.; Madhusudhan, T.; Bachhuka, A.; Kosanovic, D.; Weissmann, N.; Krüger, K.; Ergün, S. Chronic Obstructive Pulmonary Disease and the Cardiovascular System: Vascular Repair and Regeneration as a Therapeutic Target. Front. Cardiovasc. Med. 2021, 8, 649512.
- Ebina, M.; Shimizukawa, M.; Shibata, N.; Kimura, Y.; Suzuki, T.; Endo, M.; Sasano, H.; Kondo, T.; Nukiwa, T. Heterogeneous increase in CD34-positive alveolar capillaries in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2004, 169, 1203–1208.
- Simler, N.R.; Brenchley, P.E.; Horrocks, A.W.; Greaves, S.M.; Hasleton, P.S.; Egan, J.J. Angiogenic cytokines in patients with idiopathic interstitial pneumonia. Thorax 2004, 59, 581–585.
- Cosgrove, G.P.; Brown, K.K.; Schiemann, W.P.; Serls, A.E.; Parr, J.E.; Geraci, M.W.; Schwarz, M.I.; Cool, C.D.; Worthen, G.S. Pigment epithelium-derived factor in idiopathic pulmonary fibrosis: A role in aberrant angiogenesis. Am. J. Respir. Crit. Care Med. 2004, 170, 242–251.
- Liu, X.; Qin, X.; Qin, H.; Jia, C.; Yuan, Y.; Sun, T.; Chen, B.; Chen, C.; Zhang, H. Characterization of the heterogeneity of endothelial cells in bleomycin-induced lung fibrosis using single-cell RNA sequencing. Angiogenesis 2021, 24, 809–821.