Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Biochemistry & Molecular Biology
Red blood cells (RBC) are the most abundant cell in the human body, with a central role in oxygen transport and its delivery to tissues. The relative simplicity of RBCs, as historically perceived, has attracted multiple efforts to leverage them as a model of simplified human cell metabolism. Indeed, RBCs exclusively rely on glycolysis (Embden–Meyerhof–Parnas pathway) to generate high-energy phosphate compounds, such as adenosine triphosphate (ATP)—whose main source in other cells is oxidative phosphorylation in mitochondria.
- red blood cell
- erythrocyte
- transfusion medicine
1. Red Blood Cell Metabolism: The Central Role of Oxygen
By recent estimates, the adult human body contains ~30 trillion cells [1,2]. Approximately 90% of these cells are derived from the hematopoietic lineage, of which the overwhelming majority are red blood cells (RBCs) [3]. The 25 trillion circulating RBCs in an adult account for ~83% of all host cells, making RBCs a type of circulating organ critical for human health [4]. The stability of the number of circulating RBCs is ensured by a delicate equilibrium between de novo erythropoiesis and erythrophagocytosis by splenic and hepatic macrophages, which recycle RBC contents, especially iron, proteins and lipids [5].
The evolution of human RBCs has maximized their capacity to transport and deliver oxygen to tissues via progressive loss of nuclei and organelles during the maturation of erythroid precursors to reticulocytes and, ultimately, mature discocytic RBCs [6]. As a result of this process, each mature RBC contains ~250–270 million copies of hemoglobin [7], with hemoglobin accounting for ~98% of the cytosolic proteome and 92% of the total proteome [8]. At full oxygen saturation, RBCs can theoretically carry up to 1 billion molecules of oxygen/cell, a function that is facilitated by the presence of all mature RBCs combined with ~2.6 g of iron (66% of the total body iron) [4]. Fenton and Haber–Weiss chemistry constantly generate the formation of hydrogen peroxide and reactive oxygen species [9,10], making the RBC’s 120 days circulatory lifespan a struggle against the oxidation [11] of proteins (especially redox-sensitive functional residues in hemoglobin such as C93 and H92 of the beta chain [12]), small molecule metabolites [13] and lipids [14]. Every day, 0.2 trillion RBCs are removed from the bloodstream and replaced by de novo erythropoiesis (2 million RBCs are produced per second [15]), accounting for ~40% of the total body mass turnover despite the small mass of each RBC (in the range of 100–300 pg) [3].
The relative simplicity of RBCs, as historically perceived, has attracted multiple efforts to leverage them as a model of simplified human cell metabolism. Indeed, RBCs exclusively rely on glycolysis (Embden–Meyerhof–Parnas pathway) to generate high-energy phosphate compounds, such as adenosine triphosphate (ATP)—whose main source in other cells is oxidative phosphorylation in mitochondria. ATP and its guanosine triphosphate equivalent GTP substantially fuel all key processes in mature RBCs, including: hemoglobin allostery [16,17] to metabolism [18], from proton pumps [19] to membrane integrity by phosphorylating structural proteins [20], from protein stabilization via fueling of transglutaminase 2 [21] to cellular mechanics [22], from cytoskeletal actin polymerization [23] to vesiculation [24], from membrane lipid symmetry by fueling phosphatidylserine flippases [25] to proteasomal activity to remove damaged proteins [26,27,28]. Ultimately, the energy-depleted erythrocyte is rapidly lost from the bloodstream [29] via intra- or, more commonly, extravascular hemolysis through splenic sequestration and erythrophagocytosis.
Two critical functional RBC pathways branch from glycolysis: the Rapoport–Luebering shunt, which generates 2,3-diphosphoglycerate (DPG), and the pentose phosphate pathway (hexose monophosphate shunt), which generates ribose phosphate and, importantly, reduced nicotinamide adenine dinucleotide phosphate (NADPH). DPG is a critical allosteric modulator of hemoglobin, promoting oxygen release from hemoglobin to counteract hypoxia (e.g., at high-altitude [30] or from hemorrhage [31]). NADPH fuels multiple antioxidant processes in RBCs [32]: (i) it is essential for the reduction in oxidized glutathione by glutathione reductases; (ii) it directly or indirectly fuels glutathione peroxidase 4 [33], catalase, peroxiredoxins [34], glutaredoxins, the thioredoxin reductase system, biliverdin reductase B [35], the ascorbate-tocopherol axis [36] and other diaphorases such as NADPH-dependent quinone oxidoreductases (NQO1) [37]. NAD(P)H-dependent methemoglobin reductases are also essential for converting (auto-oxidized) ferric hemoglobin iron back to its ferrous state. The C93 residue of hemoglobin beta also participates in antioxidant systems by buffering free glutathione [38], participating in recycling oxidized peroxiredoxin 2 [39], and contributing to nitrite reduction [40] (resulting in the pathological generation of methemoglobin in the setting of nitrite poisoning [41]). Owing to its role in redox chemistry, it has also been proposed that hemoglobin may serve as a murzyme (i.e., a redox enzyme working along the principles of the Murburn concept), thereby contributing to ATP synthesis [42].
The rate-limiting enzyme of the pentose phosphate pathway is glucose 6-phosphate dehydrogenase (G6PD). G6PD is encoded by a gene on chromosome X. Mutations of this gene are found in >500 million people around the world, with >200 G6PD mutations known in humans [43]. Individuals carrying such mutations typically present with a significant loss of enzymatic activity, ranging from <1% in the most severe forms (e.g., the Mediterranean variant-S188F) [44,45] to <10% in the common African variant (V68M); the latter is extremely common in some metropolitan areas such as New York, especially in African American communities (~13% prevalence) [46]. In China, the six most common mutations account for ~90% of G6PD-deficient alleles, with an overall national prevalence of ~2.10% [47]. Human and mouse RBCs carrying these mutations are extremely susceptible to hemolysis following oxidant insults [48,49]. As in the case of hemoglobinopathies, such as sickle cell trait [50] and beta-thalassemia [51], positive selection for these mutations in human populations is thought to be associated with the selective pressure by malaria infections in the Mediterranean and South East Asia areas, with considerable overlap between the incidence of G6PD deficiency and malaria-endemic regions [52]. Protection against mild malaria infection is also observed in heterozygous G6PD deficient females [52]. G6PD deficiency, sickle cell trait (or disease) and beta-thalassemia (minor or major) are all associated with RBC metabolic reprogramming consistent with increased susceptibility to oxidant stress-induced hemolysis (e.g., by quinone antimalarials and sulfa drugs in the setting of G6PD deficiency).
The regulation of glycolysis by oxidant stress to functional thiols in rate-limiting enzymes, including glyceraldehyde 3-phosphate dehydrogenase (GAPDH) at cysteine residues 152 and 156 [53] and pyruvate kinase [54,55], provides a strategy to constrain metabolic fluxes through late glycolysis when oxidant stress is high while redirecting glucose oxidation to the pentose phosphate pathway to produce reducing equivalents that counteract this stress. Similarly, the most abundant RBC membrane protein, band 3 (or, equivalently, anion exchanger 1—AE1) has a very acidic N-terminus cytosolic domain that can serve as an inhibitory docking site for glycolytic enzymes at high oxygen saturation; in contrast, at low oxygen saturation deoxyhemoglobin outcompetes the glycolytic enzymes, displacing them from the membrane and boosting glycolysis to stimulate ATP and DPG production in the face of hypoxia [56,57]. Prolonged, unmitigated oxidant stress, such as during refrigerated RBC storage under blood bank conditions, promotes proteolytic or reactive oxygen species (ROS)-triggered proteolysis of the N-terminus of band 3, ultimately causing the loss of this RBC oxygen-dependent metabolic modulation pathway [58,59,60]. In addition, genetic mutations of the N-terminus region of band 3 are associated with severe hemolysis and necessitate lifelong transfusion [59].
2. RBC Metabolism beyond Glycolysis
Over the last 50 years, almost all computational efforts to simulate the RBC metabolome ex vivo were limited to the pathways described above [61,62,63]. The recent implementation of omics technologies to study RBCs has revealed an unanticipated complexity of the erythrocyte proteome, now counting ~2500–3000 unique proteins; indeed, functional metabolic tracing experiments suggest that even this list may be incomplete [8,59,64,65,66,67,68,69]. Leveraging these recent datasets, systems biology experts are redrawing connectivity maps of the human RBC metabolome (Figure 1), of relevance for basic science and translational applications [70,71]. With >77 active transporters, circulating RBCs can take up and release many metabolites from peripheral tissues, making RBCs a unique window into system health [72]. From creatinine and carnitine (as markers of renal function [73]) to conjugated bile acids (from the gut microbiome [74]), from transamination products (e.g., alanine, glutamate, aspartate) to neurotransmitters (e.g., serotonin, dopamine, acetylcholine), RBCs can directly and indirectly participate in systems metabolism throughout the body.
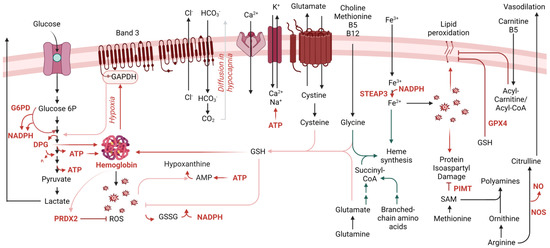
Figure 1. Overview of the main metabolic pathways relevant to RBC physiology during aging in vivo and in vitro.
During erythropoiesis, amino acid metabolism (e.g., glycine, glutamine, branched-chain amino acids) is essential to heme synthesis as both direct and indirect substrates (e.g., succinyl-CoA from glutaminolysis and branched-chain amino acid catabolism). This process concomitantly releases ammonium, making erythropoiesis and de novo glutamine synthesis two of the leading pathways contributing to ammonium homeostasis in humans [75]. Genetic defects of rate-limiting genes in the branched-chain amino acid catabolism (e.g., in propionic [76] or methylmalonic acidemias) are associated with defects in erythropoiesis [77]. Along with cysteine, both glycine and glutamine-derived glutamate contribute to the synthesis of the glutathione tripeptide. The exchange of cystine—a cysteine disulfide—to glutamate is a critical regulator of a process previously referred to as eryptosis [78] and, more recently, recognized as ferroptosis [79] owing to its expanded relevance in other cells besides RBCs.
When considering erythropoiesis, one-carbon metabolism is one of the first pathways that is referenced. Folates and methyl group donors such as methionine, choline and betaine—along with B6, B12 and B5 cofactors [80,81]—are essential for de novo purine nucleotide synthesis to support proliferation, as well as for generating glycine to contribute to heme synthesis described above. However, methyl-group donors also participate in RBC redox homeostasis and its dysregulation, such as in the context of folate dietary deficiency or excess [81]. This is also relevant in diseases associated with alterations of this pathway such as homocystinuria [82] or Down syndrome [83], owing to gene dosage of cystathionine beta synthase resulting in a “folate trap”-like phenotype [84]. These conditions present with macrocytic or megaloblastic anemia [82]. In addition, in keeping with the central role of oxidant stress in the economy of RBC metabolism, methionine uptake and consumption in RBCs fuel a pathway of isoaspartyl protein damage repair. As a hallmark of RBC aging in vivo [85], deamidation of asparagine residues in key structural membrane proteins and glycolytic enzymes [86,87,88,89,90,91,92] alters the protein backbone, with important structural/functional implications. Since RBCs cannot replace damaged components by de novo protein synthesis, repairing this damage is essential to their survival. The methylation of deamidated residues favors the formation of a succinimide intermediate, ultimately promoting the rescue of the protein backbone structure in 15–30% of the cases, a reaction catalyzed by the enzyme “protein isoaspartyl o-methyltransferase” (PIMT). Increased uptake of methyl donors such as methionine is observed in RBCs in response to oxidative challenges (e.g., incubation with hydrogen peroxide [93], refrigerated storage under blood bank conditions [92]). Thus, one can speculate that RBC uptake and consumption of methyl-group donors (e.g., methionine, choline) could compete with other tissues where these substrates are used to fuel epigenetic regulatory mechanisms, such as methylation of proteins (e.g., histones), RNA (N6-methyladenosine) and DNA (CpG islands), making RBCs an indirect player in systemic long-term responses to oxidant stress events.
In response to oxidant stress, ATP synthesis is reduced because of the redox sensitivity of glycolytic enzymes and the band 3-dependent mechanism, as described above. In this case, ATP breaks down into lower energy phosphate compounds, ADP and AMP, the latter being prone to deamination to IMP by RBC-specific AMP deaminase 3 [94]. Phosphoribolysis of IMP releases hypoxanthine, a substrate for xanthine oxidase to generate xanthine and urate, with concomitant production of hydrogen peroxide [94]. The presence of an active xanthine oxidase in mature RBCs was reported and challenged [95]. Exogenous urate is a potent antioxidant in mature RBCs [96]. Similarly, the breakdown of AMP into adenosine, with adenosine oxidation by adenosine deaminase was reported as contributing to hypoxanthine accumulation [97]. The recycling of hypoxanthine by X-linked hypoxanthine guanosine phosphoribosyltransferase (HPRT) preserves IMP/GMP homeostasis in RBCs. Lesch Nyhan syndrome patients exhibit genetic mutations of this enzyme and present with a range of clinical manifestations, including macrocytic anemia [98]. Purine homeostasis is relevant to RBC homeostasis for additional reasons beyond those described above. For example, ATP release by RBCs has regulatory effects on endothelial cells and on the RBCs themselves, as a type of autocrine signaling. The breakdown of extracellular ATP to ADP, AMP and adenosine by ectonucleotidases (e.g., CD38) generates agonists of P2Y receptors on endothelial cells, but adenosine can also be imported by equilibrative nucleotide transporters (ENT1) or stimulate adenosine receptors (e.g., ADORA2b). Both of these pathways contribute to RBC responses to hypoxia, the former, for example, by limiting circulating adenosine, a process that is counteracted by irreversible ENT1 degradation to facilitate acclimatization to high altitude hypoxia upon reascent [99]. In addition, ADORA2b transduces an intracellular signaling pathway that activates downstream protein kinase A and AMP-dependent kinase (AMPK), both contributing to metabolic reprogramming; for example, via phosphorylation-mediated activation of bisphosphoglycerate kinase [97,100].
Recent applications of unsupervised metabolomics and lipidomics approaches identified a sphingolipid sphingosine 1-phosphate (S1P) as a relevant metabolite regulating RBC responses to hypoxia [101]. For example, in response to high altitude-induced hypoxia [101] S1P can bind to deoxyhemoglobin following its stabilization by DPG, further promoting oxygen off-loading [102]. By further stabilizing deoxyhemoglobin, S1P also contributes to energy metabolism by promoting the release of glycolytic enzymes from band 3 into the cytosol, with subsequent activation of glycolysis at the expense of the pentose phosphate pathway. Although this adaptation is beneficial in hypoxia, it is deleterious when RBCs are challenged by oxidant stress, such as during refrigerated storage in the blood bank [103] or in sickle cell disease, where deoxy-sickle hemoglobin stabilization further promotes its crystallization [102].
Arginine metabolism in mature RBCs was found to be more complex than anticipated, despite the absence of mitochondria, where a subset of critical reactions in the urea cycle are known to occur. For example, RBCs contain high levels of arginase 1 [113], which converts arginine to ornithine, a precursor of polyamines via ornithine decarboxylase. Although this pathway is critical in erythropoiesis, owing to the role of polyamines in regulating the intracellular pH of hematopoietic precursors [114], in adult RBCs this pathway has been associated with cellular responses to iron-deficient anemia and abiotic stresses, such as exposure to radiation [115]. Arginine metabolism cross-talks with heme synthesis, glutathione homeostasis and one-carbon metabolism, in that polyamine synthesis and creatine synthesis are both affected in human RBCs by factors such as sex and age [48], and compete for rate-limiting substrates for the generation of each product. During the last decade, RBCs were found to harbor a functional nitric oxide synthase [116], which converts arginine to citrulline and concomitantly generates nitric oxide, a potent vasodilator with a crucial role in the regulation of endothelial cells and related vascular function [117,118].
As already mentioned above, carboxylic acid intermediates of the Krebs cycle, such as succinyl-CoA participate in erythropoiesis by fueling heme synthesis. In addition, small molecule dicarboxylates in this pathway (e.g., succinate, fumarate, malate) sustain erythropoiesis by the mechanism of stabilization of the Hypoxia Inducible Factor 1alpha upon exposure to hypoxia, which otherwise is degraded following hydroxylation by alpha-ketoglutarate-dependent prolyl hydroxylases [119]. The conversion of alpha-ketoglutarate to 2-hydroxyglutarate can occur under hypoxic conditions by non-canonical lactate dehydrogenase activity [120], and testosterone-induced stimulation of erythropoiesis [121]. The absence of mitochondria in healthy mature RBCs originally led the field to believe that minimal carboxylic acid metabolism would occur in this cell system. However, proteomics recently identified functional cytosolic isoforms of acetyl-CoA ligase, isocitrate dehydrogenase 1, malate dehydrogenase 1 and malic enzyme 1; thus, catabolism of pyruvate and citrate can occur in mature RBCs [122,123] via a series of reactions that can fuel the alternative generation of reducing equivalents (e.g., NADPH and NADH), especially in hypoxia [124]. The activation of these pathways may be especially beneficial as a partial compensatory mechanism in the face of genetic aberrations leading to a dysfunctional pentose phosphate pathway, such as G6PD deficiency [46,125].
Although healthy mature RBCs are indeed devoid of mitochondria, recent evidence unequivocally documented the presence of up to 6–7 mitochondria per cell in mature RBCs from sickle cell patients [126,127,128,129]. Similar observations have been reported in the context of other pathophysiological conditions, such as systemic lupus erythematosus [130] and Rett syndrome [131,132]. Whether and to what extent these organelles still function is incompletely understood, though their retention might result from a defect in mitophagy or the ubiquitin-proteasome system [126,127,132] Nonetheless, the presence of metabolically active mitochondria in mature RBCs could induce oxygen consumption and consequently oxidant stress through ROS generation [128]. The induction of reticulocytosis or heterogeneous retention of mitochondria is associated with alloimmunization in a murine model of transfusion [133], which could be relevant to blood donors with heterogeneous mitochondrial DNA content in circulating erythroid cells and recipients with inflammatory conditions such as systemic lupus erythematosus [131]. The relevance of this phenomenon in RBC aging in vivo—to the extent this may result from/contribute to processes of age-related aberrant erythropoiesis and age-related comorbidities remains to be determined. Nonetheless, one may speculate that intra- or extra-vascular hemolysis of mitochondria-containing RBCs may release prokaryotic-like RNA and DNA into the circulation or in phagocytic macrophages, thereby triggering cGAS-STING-Interferon responses [130,134], ultimately driving interferon-mediated inflammatory complications in sickle cell patients, lupus patients and in other conditions in which interferonopathies and hematological anomalies are observed (e.g., Down syndrome) [135].
This entry is adapted from the peer-reviewed paper 10.3390/metabo13070793
This entry is offline, you can click here to edit this entry!