2. SLRPs Modulate Cancer Cell Adhesion, Migration, and Invasion
Migration and adhesion are cell functions that are correlated with invasion and directly involved in embryo tissue development, tissue homeostasis, and regeneration. Invasion cell properties are also needed during the penetration of biological barriers that lead to metastasis [
101]. During the epithelial-to-mesenchymal transition of cells in tumors, cells experience loss of intrinsic polarity, resulting in the loosening of cell-to-cell junctions. The latter process is supported by reorganizing the cell cytoskeleton and actin-based cell motility activation [
102]. Cells that partially retain their polarity can form local adhesions and still attach to the ECM and perform cell body translocation [
102]. Furthermore, cells with no polarity are amoeboid-like and can chemotactically move towards stimuli due to increased migration capacity and weakly attaching to the ECM [
102].
2.1. Biglycan
Biglycan was described to modulate tumor cell migration and invasion capabilities. In the case of endometrial cancer, biglycan regulated cell invasion and migration cell capacity in vitro and experiments using a xenograft model [
103]. A recent study showed that the sponging of miR-320a upregulates biglycan expression to support head and neck squamous cell carcinoma (HNSCC) progression by inducing EMT [
104]. In this case, HNSCC cells exhibited increased migration and invasion, as well as enhanced expression of EMT markers, such as N-cadherin and vimentin.
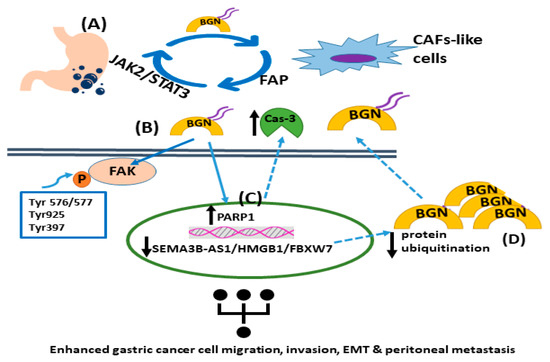
In addition, biglycan, via in vitro and in vivo models of gastric cancer, was shown to increase tumor cell invasion and metastasis through activation of FAK signaling via specific phosphorylation at Tyr576/577, Tyr925, and Tyr397 [
105]. Wu et al. recently presented a positive feedback loop that involved biglycan-, FAP-, and STAT3-mediated interactions between tumor cells and mesothelial cells that contribute to peritoneal metastasis of gastric cancer [
106]. Specifically, this study identified an intricate loop through which gastric cancer cell-derived biglycan facilitates mesenchymal cell transformation to CAF-like cells, activating TLR signaling. In turn, CAF-like cells secrete fibroblast activation protein (FAP) to promote gastric cancer invasion, migration, and EMT. Moreover, FAP triggers the JAK2/STAT3 signaling pathway in gastric cancer cells, facilitating their biglycan expression to initiate the vicious circle of cancer progression [
106]. In a separate study, the SEMA3B-AS1/HMGB1/FBXW7 signaling axis was shown to be decreased in gastric cancer cells, which enhanced the peritoneal metastasis of gastric cancer by modulating biglycan protein ubiquitination and concomitant degradation, resulting in higher biglycan levels [
107]. Likewise, biglycan-deficient gastric cancer cells display attenuated migration compared to biglycan-expressing cells. Indeed, biglycan knockout gastric cancer cells have enhanced PARP1 levels and caspase-3 cleavage with a concomitant attenuation of mesenchymal marker expression. The in vitro and in vivo data were verified via patient biopsies [
108]. These findings are supported by a study that showed that celastrol, which is a necroptosis inducer, triggered RIP1/RIP3/MLKL signaling pathways and attenuated pro-inflammatory cytokine release by targeting biglycan in gastric cancer cells [
109]. The mechanisms of biglycan action in gastric cancer are presented in
Figure 1.
Figure 1. The roles of biglycan in gastric tumor carcinogenesis. (A) Gastric cancer cell-derived biglycan enhances the transformation of mesenchymal cells into CAF-like cells, which activates TLR signaling. In turn, CAF-like cells secrete fibroblast activation protein (FAP), promoting gastric cancer migration, invasion, and EMT. FAP initiates the JAK2/STAT3 signaling pathway in gastric cancer cells, increasing their biglycan expression. (B) Biglycan stimulates gastric cancer cell invasion and metastasis by activating FAK signaling via specific phosphorylation at Tyr576/577, Tyr925, and Tyr397. (C) Biglycan decreases PARP1 levels and caspase-3 cleavage with a concomitant upregulation of mesenchymal markers. (D) The SEMA3B-AS1/HMGB1/FBXW7 signaling axis is downregulated in gastric cancer cells, which facilitates the peritoneal metastasis of gastric cancer by decreasing biglycan protein ubiquitination and concomitant degradation, leading to higher biglycan levels.
Recently, biglycan was shown to enhance specific breast cancer stem cells’ (BCSC) migration and invasion. In this study, biglycan-deficient BCSCs exhibited decreased metabolism, reduced proportions, and attenuated ability to form tumor spheroids. Mechanistically, these cells demonstrated a decrease in NFκB transcription factor activity and downregulated phospho-IκB levels [
110]. Moreover, ALDH
+ and CD29
hi CD61
+ BCSCs deficiency in biglycan were characterized by reduced metastatic ability. Manupati et al. suggest that biglycan could be a therapeutic target in BCSCs, making it helpful in developing novel strategies. Furthermore, biglycan was found to reduce HER-2/neu-transformed cells’ tumor properties, which were shown to be inversely correlated with PKC signaling [
111].
Moreover, K-RAS-transformed colorectal cancer cells, which overexpress biglycan exhibit lower migratory properties [
112]. These authors suggest that K-RAS-dependent malignancies could be treated via the upregulation of biglycan.
Datsis et al. investigated the role of biglycan in mesenchymal-originating tumors [
113]. A co-operative mechanism of parathormone (1–34) and FGF-2 action was demonstrated using osteosarcoma cell lines to enhance biglycan expression and increase cell migration [
113].
In contrast, this SLRP was determined to affect, in colorectal cancer, the desmoplastic reaction, inhibiting migration and invasion of these tumor cells in 2D and 3D coculture systems [
114]. In cancer, the stroma tends to produce, in an unorganized fashion, fibrous tissue that consist mainly of collagen [
115]. This response is named desmoplasia, and the fibrotic tissue is primarily synthesized by cancer-associated fibroblasts [
115,
116]. Another study on cell motility and biglycan in pancreatic cancer cells explains that the RAC1B/SMAD3/biglycan signaling axis attenuated migration [
117]. These data demonstrate a complex pattern of biglycan actions, highlighting the need for more detailed studies.
2.2. Lumican
Cells’ 2D and 3D migration are facilitated by forming locomotory cytoplasmic (filopodia, pseudopodia, lamellipodia) or plasma membrane (invadopodia) protrusions that adhere to ECM components with the help of protein degradation molecules, like matrix metalloproteinases (MMPs) [
118,
119,
120]. Invadopodia and lamellipodia formation were shown to be inhibited by lumican in prostate cells [
121]. Experiments that plated prostate cancer cells on a lumican substrate resulted in the inhibition of lamellipodia formation via reduced rearrangement of ZO-1, keratin 8/18, integrin β1, and MT1-MMP. In addition, invadopodia inhibition was observed through disruption of α-smooth muscle actin, cortactin, and N-WASP [
121]. Similarly, attenuation of invadopodia formation by lumican was shown in melanoma cells [
122]. Furthermore, lumican was proven to affect the Snail-dependent MMP-14 action in melanoma cells; however, this process did not occur in the HT-29 colon adenocarcinoma cell model [
123]. Furthermore, lumican reduced melanoma migration capacity via core protein binding to α2β1 integrin [
124].
This SLRP modulates the expression of ECM molecules and reduces MMP-releasing invadopodia to inhibit melanoma lung metastasis in vivo and cell invasion, as Karamanou et al. (2021) recently showed. Also, lumican in breast cancer modulated cell morphology, while EMT decreased the expression of ECM regulators, like MMPs, and inhibited cell functions, such as migration and invasion [
125].
In contrast, inhibition of lumican expression reduced bladder cancer cells’ migration capacity through MAPK signaling pathway activation [
126]. In a separate study, lumican enhanced the adhesion of lung cancer cells to different ECM components and increased their in vitro cell migration, invasion, and osteogenic metastasis [
127]. Furthermore, lumican overexpression increased podosome-like protrusions’ formation and migration in human colon cancer [
128]. Experiments in an osteosarcoma cell model showed that lumican aided cell migration and chemotactic response to fibronectin, as well as facilitating the TGFβ2-dependent adhesion capacity of the same cells onto fibronectin [
44,
52,
129]. Moreover, a FOX-3-dependent increase in lumican expression facilitated the migration of aggressive neuroblastoma cells [
130].
The altered stroma ECM could significantly regulate growth, migration, and cancer spread [
115]. Patients with lumican-positive stains of cancer stroma in pancreatic cancer presented shorter survival rates than those with lumican-negative stroma [
131]. The different roles of lumican are summarized in
Table 1.
Table 1. Lumican’s actions in cancer adhesion, migration, and invasion.
Biglycan and lumican were proven to exert cancer-type specific roles in these cancer cell functions; thus, further investigation should aim to complete the detailed description of respective SLRPs’ mechanisms of action.
3. SLRPs Affect Tumor Cell Growth and Cell Cycle Regulation
One of the main steps involved in tumorigenesis is uncontrolled and sustainable cell proliferation. The deregulated function of cancer cells leads to the continuous release of growth-promoting signals, such as growth factor production that binds their tyrosine kinase activity receptors and activates their signaling, finally enhancing malignant transformation and tumor progression [
132]. The interaction of SLRPs with vital signaling molecules on the cell surface or in the microenvironment is well-established [
2]. Biglycan and lumican can positively or negatively affect malignant cell growth, depending on the type of cancer and their role in this particular tumor’s behavior.
3.1. Biglycan
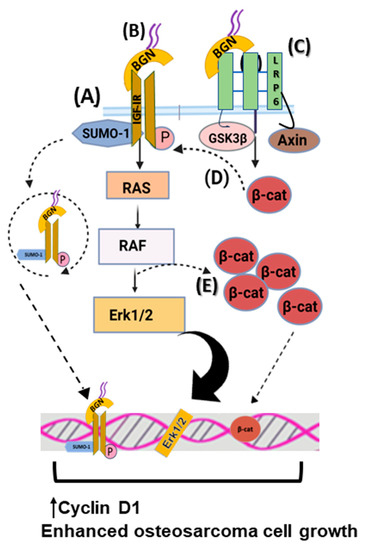
Biglycan is reported to significantly stimulate mesenchymal-derived tumor cell growth through complex regulating mechanisms. Thus, biglycan activates the insulin-like growth factor receptor I (IGF-IR) and Wnt/β-catenin signaling cascade, enhancing osteosarcoma growth. Biglycan-deficient cells show reduced activation of ERK1/2 and attenuated expression of Cyclin D1, which is a gene related to cell-cycle control [
23]. Moreover, biglycan is shown to bind the IGF-IR receptor, enhancing its sumoylation and translocation to the nucleus. The nuclear IGF-IR has a transcriptional role, affecting the expression of target genes, including Cyclin D1 [
25]. Moreover, computational studies reveal that biglycan changes the conformation of Wnt co-receptor low-density lipoprotein receptor-related protein 6 (LRP6) and promotes its ability to interact with signaling molecules. Biglycan binding to LRP6 disrupts the formation of the β-catenin degradation complex, resulting in β-catenin nuclear translocation and transcriptional upregulation of target genes, including Cyclin D1 [
23,
25]. Moreover, β-catenin was shown to co-localize with IGF-IR, prolonging its activation and downstream signaling. Previously, biglycan was shown to stimulate canonical Wnt signaling through binding to LRP6, resulting in enhanced bone fracture healing [
133]. Collectively, these data suggest that by hijacking biglycan growth-promoting signaling, tumors can enhance their growth. The effects of biglycan in regulating osteosarcoma cell growth are depicted in
Figure 2.
Figure 2. Biglycan upregulates osteosarcoma cell growth. (A) Biglycan binds to IGF-IR, enhancing its activation, subsequent sumoylation, and translocation to the nucleus. Nuclear IGF-IR regulates the transcription of target genes, including Cyclin D1, thus increasing osteosarcoma cell growth. (B) Upon biglycan binding to IGF-IR and its phosphorylation, downstream RAS/RAF/Erk1/2 signaling cascade is activated, resulting in transcriptional regulation that enhances osteosarcoma cell growth. (C) Biglycan binds to LRP6 and disrupts the formation of the β-catenin degradation complex, resulting in β-catenin nuclear translocation and transcriptional regulation that promotes osteosarcoma cell growth. (D) β-catenin co-localizes with IGF-IR, prolonging its activation and downstream signaling, which is correlated with increased osteosarcoma cell proliferation. (E) Activated Erk1/2 and β-catenin co-localize to facilitate β-catenin intracellular pool.
Notably, the LRP6/biglycan axis was, likewise, detected in glioblastoma cells, suggesting that it could also be relevant in advancing neural exoderm-derived malignancies [
134]. Specifically, in a syngeneic glioblastoma mouse model, biglycan expression was increased in areas infiltrated with brain tumor-initiating cells. Treating brain tumor-initiating cells via exogenous biglycan or its overexpression increased their growth through biglycan binding to LRP6 and activation of Wnt/β-catenin signaling [
134].
Biglycan, likewise, promotes the progression of epithelial-derived colorectal cancer. HCT116 biglycan-deficient colon cancer cells exhibit altered expression of cell-cycle regulator genes and lower proliferation rates, as well as cell-cycle arrest at the G0/G1 phase [
135,
136]. As hedgehog signaling, which results in biglycan overexpression, is overactivated in colon cancer, this process leads to enhanced tumor cell growth in colony formation and xenograft assays [
137].
In addition to mainly growth-promoting effects, some studies show that biglycan inhibits cancer cell growth. For example, in pancreatic cancer, biglycan overexpression leads to G1 arrest and attenuates cell proliferation [
138]. In addition, human urothelial carcinoma cells (J82 cells) that were deficient in biglycan exhibited enhanced proliferation and a decreased ability to form xenografts. Furthermore, treating J82 cells with recombinant biglycan in vitro strongly decreased their proliferation, confirming biglycan’s antiproliferative effect [
86].
3.2. Lumican
It is well established that lumican controls tumor cells’ proliferation in a cancer-type-dependent manner. Thus, lumican’s positive correlation with gastric cancer is reported. CAFs in gastric cancer express lumican, which promotes the activation of the integrin β1-FAK signaling pathway, resulting in increased proliferation of cancer cells and tumor progression. Knockout of lumican reduces cell growth and metastasis in vivo [
139]. Furthermore, lumican enhances cell proliferation in lung cancer cell lines, as lumican deficiency affects mitotic spindle and midbody formation, leading to chromosome mis-segregation and increased chromosome instability. Lumican-deficient H460 and A549 in non-small lung cancer cell lines exhibit a prolonged doubling time and stunted cell growth [
140].
Regarding chondrosarcoma, lumican has a major role in cell proliferation mechanisms. A prior study demonstrated that lumican is the most abundantly expressed SLRP in HTB94 chondrosarcoma cells, and its deficiency is connected to reduced phosphorylation levels of IGF-IR and extracellular signal-regulated kinase ERK1/2 activation, which is a crucial signaling pathway for IGF-I-dependent HTB94 cell growth [
26]. Lumican also modifies cancer response to chemotherapeutics [
141]. A novel quinoline compound 91b1, which has strong anticancer effects in different cancer cell line models and in vivo, downregulates lumican mRNA levels, which are overexpressed in many cancers. Zhou et al. correlate lumican downregulation with suppressed cell proliferation and modulated cell cycle progression [
142].
In osteosarcoma, lumican expression did not affect the growth of the aggressive MG63 cells, while attenuated non-aggressive Saos-2 cells grew through the TGF-β2 signaling cascade, affecting the bioavailability of Smad 2 activators [
44,
52]. Notably, in pancreatic ductal adenocarcinoma (PDAC) cancer, lumican, which is expressed by stromal cells, has an antiproliferative effect. Indeed, lumican’s expression attenuates the expression and activation levels of epithelial growth factor receptor (EGFR), leading to subsequent downregulation of Akt kinase activity and reduced tumor growth. Li et al. also demonstrated that PDAC cell treatment with exogenous lumican promotes cell-cycle arrest at G0/phase and cell-growth inhibition mediated via decreased ERK1/2 activation and increased p38 phosphorylation in PDAC cells [
143,
144]. Moreover, lumican inhibits tumor growth in melanoma mouse models by sensitizing the matrix microenvironment to ECM-targeted therapy [
145]. These data suggest that stroma-derived lumican negatively affects cancer cell functions.