Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Medicine, Research & Experimental
Amyotrophic lateral sclerosis (ALS) is a severe and incurable neurodegenerative disease characterized by the progressive death of motor neurons, leading to paralysis and death. It is a rare disease characterized by high patient-to-patient heterogeneity, which makes its study arduous and complex. Extracellular vesicles (EVs) have emerged as important players in the development of ALS.
- amyotrophic lateral sclerosis
- neurodegenerative diseases
- extracellular vesicles
1. The Current State-of-the-Art Research of ALS
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease. Described for the first time in the 19th century by Charcot, ALS is characterized by the degeneration of lower (spinal and bulbar) and upper (corticospinal) motor neurons [1]. The selective loss of motor neurons (MNs) in the primary motor cortex, brainstem, and spinal cord progressively leads to severe effects, such as loss of motor control, paralysis, and death. Death usually occurs due to respiratory failure. Approximately half of the patients with ALS show impairments in cognitive function and behavior, with 5–25% of patients developing frontotemporal dementia (FTD) [2,3,4], which is an uncommon type of dementia characterized by changes in the frontal and temporal lobes.
ALS is considered a rare disease, with an incidence that ranges between 0.6 and 3.8 persons out of 100,000 and a prevalence between 4.1 and 8.4 per 100,000 individuals, with an average age of onset between 51 and 66 years [5]. The life expectancy of patients with ALS is short, ranging between 24 and 50 months. However, approximately 10% of patients manage to live for more than 10 years [5], a fact that reflects the high patient-to-patient phenotypic variability that characterizes ALS. The triggering elements of the disease remain unknown, although genetic causes can be attributed to several individuals. Some studies point to the possibility of an oligogenic or polygenic nature, as mutations in two or more genes may be required for the disease [6,7]. People with a history of ALS in their family and those carrying ALS-related genes are more likely to develop the disease (familial, fALS), representing 5–10% of all cases. For the remaining 90% to 95%, the illness can occur spontaneously, without a family history (sporadic ALS) [6], and still be linked to ALS-related genes. Currently, ALS is difficult to diagnose due to the absence of a test that can solely lead to its identification unless it is a familial form. In the absence of a family history, a battery of examinations is often performed to exclude other possible pathologies. Currently, ALS remains cureless, and the available treatments are sparse and mostly palliative. Two approved medications are currently prescribed to patients, Riluzole and Edaravone, with the latter only being approved in some countries. However, they only present small benefits in delaying ALS progression, usually only by a few months [8]. Therefore, the discovery of new and effective drugs is of utmost importance.
1.1. Risk Factors for ALS Onset and Progression
The likelihood of developing ALS and its progression are influenced by numerous factors, including genetic and non-genetic origins. One important non-genetic factor is age, as individuals who develop ALS in early adulthood tend to experience slower disease progression rates [9,10]. Another factor is gender, with men being about 1.3 times more likely to develop ALS than women and earlier in life [11]. Gender also plays a role in ALS onset type, with spinal onset more common in men, while women are more likely to present bulbar onset [10]. In addition to genetic factors, exposure to certain modifiers throughout an individual’s life may also contribute to the risk of developing ALS [12]. Several environmental and lifestyle factors have been identified as potential risk factors for ALS onset, including hazardous smoking habits [13], higher lipid levels [14], prolonged exposure to pollutants [15], heavy metals [16], chemicals [17], electromagnetic fields [17], a history of electric shock [18], and head trauma [19]. Other factors linked to an increased risk of ALS include military service [20], participation in professional sports [21,22], and occupations that involve repetitive physical work [12,23,24]. However, some of these factors have been contested, owing to studies with inconclusive results [25]. These factors can eventually lead to epigenetic and genomic changes that may contribute to ALS onset, such as the occurrence of C9ORF72 (chromosome 9 open reading frame 72) somatic mutations [26]. Scientific reports have consistently indicated an interaction between the genetic and environmental risk factors. Epigenetic alterations, mostly comprising DNA methylation, were identified by screening the biofluids and postmortem brain and spinal cord tissues. In this regard, Morahan et al. (2009) reported gene and CpG island methylation in 38 differentially methylated sites in brain samples from 10 sALS patients [27]. Similarly, Figueroa-Romero et al. (2012) identified 3574 methylated genes in postmortem sALS patients’ spinal cords [28]. Cai et al. (2022) recently proposed a role for DNA methylation in ALS pathogenesis. Their study analyzed and compared the blood of 32 healthy controls with 32 sALS patients, leading to the identification of 12 differentially methylated regions (DMRs) in 12 genes and 34 differentially methylated positions (DMPs) in 13 genes. Abnormal methylation patterns were primarily associated with genes involved in the regulation of crucial cellular functions that have previously been linked to ALS, including microtubule-based movement, ATP-nucleotide binding, and neuronal apoptosis [29]. Despite research efforts to elucidate the impact of environmental and lifestyle factors on different cellular and molecular processes involved in ALS onset and progression, the exact mechanisms underlying motor neuron degeneration are still not sufficiently understood [1].
1.2. ALS Genetics and Associated Mechanisms
ALS is a highly heterogeneous disease caused by a wide array of different genes with hundreds of possible mutations [30]. Consequently, distinct fundamental cellular processes have been reported to be dysfunctional in different stages of the disease, including DNA repair mechanisms, RNA metabolism, mRNA axonal transport, protein homeostasis, protein trafficking, protein misfolding and aggregation, calcium regulation, mitochondrial function [31], redox signaling, lipid metabolism, glutamate signaling, and autophagy [32]. ALS-related gene mutations may also affect intercellular communication and function, such as neurovascular function [33,34,35], glial-related neuroinflammation [10,36,37,38], and neuron–glia interaction [39,40]. Among the several genes identified as ALS-related, some are involved in both fALS and sALS, such as TDP-43 (TAR DNA-binding protein 43), also known as TARDBP (transactive response DNA-binding protein), SOD1 (copper zinc superoxide dismutase 1), C9ORF72, and FUS (fused in sarcoma), among others [41]. Nevertheless, for 32% and 89% of patients with fALS and sALS, respectively, the mutations involved are unknown [42,43].
One of the most studied ALS-related genes is SOD1, which encodes for an important antioxidant protein, superoxide dismutase [44], responsible for converting superoxide radicals in hydrogen peroxide and oxygen [45]. Mutant SOD1 (mSOD1) alters different metabolic pathways and results in the formation of misfolded SOD1 protein aggregates and neurodegeneration [46,47]. Accordingly, mSOD1 aggregate accumulation impairs axonal transport and is neurotoxic to spinal cord MNs from the pre-symptomatic phase onwards in the ALS mice SOD1-G93A model [48]. mSOD 1 is also responsible for the alteration of the dynamic interaction between MNs and their surrounding glial cells, evoking a non-cell autonomous toxicity mechanism driven either by the promotion of the secretion of neurotoxic cytokines, through the loss of glial cells supporting properties, or both, leading to the death of MNs [49,50]. In one proposed mechanism, extracellular mSOD1 is endocytosed by microglia and activates caspase-1, leading to the upregulation of IL-1β [51,52]. IL-1β is a proinflammatory cytokine that is potentially involved in ALS neuroinflammation-related processes [53], like microgliosis and astrogliosis. In postmortem tissue samples from ALS patients, microglia are in a proinflammatory state [54] and release several cytokines, such as IL-1α and TNF-α, which induce astrocyte neurotoxicity [54]. Such evidence points to deleterious crosstalk between microglia and astrocytes, thus tracing an increased proinflammatory and neurotoxic microenvironment. Therefore, the progressive degeneration of corticospinal and spinal motor neurons may depend on their vulnerability to both mSOD1 aggregate accumulation and the effects of the surrounding glial cell dysregulation, which emphasizes the simultaneous occurrence of lower and upper MN degeneration [55].
The most commonly mutated gene in both patients with fALS and sALS is C9ORF72. The C9ORF72 gene contains 11 exons, and (GGGGCC)n is located between exons 1a and 1b. (GGGGCC)n is located in the first intron of V1 and V3 and in the promoter region of variant 2. This gene codes for a protein with the same name whose function is not fully understood but is thought to be involved in different cellular activities, such as protein transport, vesicle formation, autophagy, RNA processing, and cell signaling [56,57]. It has been suggested that C9orf72 may play a role in the autosomal and lysosomal function of macrophages and microglia through the regulation of inflammatory responses, possibly related to MN survival, relevant in ALS [58,59]. Wild-type C9orf72 forms a complex with SMCR8 (Smith–Magenis syndrome chromosomal region candidate gene 8) and WDR41 (WD40 repeat-containing protein 41) to perform these functions, including their effect on macrophages and microglia [57,60]. Because of the nature of this procedure, this function has been proposed to be affected in ALS in the presence of mutations; however, further studies are needed [57,59]. C9ORF72 mutation is not only the most common mutation in ALS but is also responsible for FTD. This mutation is reflected as an increase in the number of hexanucleotide (G4C2)n repeat expansions (HRE) in the noncoding region of C9ORF72, which results in both loss of function linked to C9ORF72 haploinsufficiency and a gain of function, resulting in the expression of abnormal bidirectionally transcribed RNAs carrying the repeat [61]. This repeat expansion leads to abnormal RNA molecule biosynthesis, which is then translated into dipeptide repeat proteins (DPRs) containing multiple copies of the specific amino acid sequence GGGGCC. DPRs, such as poly-proline-arginine (poly-PR), poly-glycine-arginine (poly-GR), and poly-glycine-alanine (poly-GA), are cytotoxic [62], accumulate in neurons [63], and may then spread to glial cells via intercellular communication [64], thus impairing protein folding and transport, inducing oxidative stress, and disrupting mitochondrial function [65]. An important player in the pathophysiology of ALS patients carrying C9ORF72 expansion is poly GA, which induces the intracellular aggregation of phosphorylated TDP-43 proteins through the impairment of TDP43 nuclear translocation and cytoplasmic mislocation [66,67]. A pathological hallmark of these patients is the presence of TDP-43 inclusions in neurons and oligodendroglial cells. The C9ORF72 gene has also recently been associated with nucleolar dysfunction [68] and DNA repair inhibition [69]. Other important cellular processes that are affected by C9ORF72 gene mutation are vesicular and protein trafficking [70]. C9ORF72 HRE was found to reduce the interaction between C9orf72 and the Rab GTPase key regulator Rab7L1, resulting in decreased extracellular vesicle (EV) release [70]. The role of C9orf72 in protein trafficking was further demonstrated in the human spinal cord of an ALS patient (with a C9ORF72-intronic repeat expansion mutation), where an increased proportion of motor neurons showed the colocalization of C9orf72 with Rab 5, Rab 7, and Rab 11 (when compared to healthy individuals), possibly resulting in the dysregulation of endosomal trafficking [70]. Interestingly, these proteins were recognized to be associated with vesicle trafficking regulation from the multivesicular bodies (MVB) to the plasma membrane, being involved, among other instances, in autophagy [70].
TARDBP, which codes for TDP-43, is another commonly mutated gene in ALS. Under normal physiological conditions, TDP-43 is primarily found in the nucleus, where it participates in the regulation of gene expression [71]. However, mutations in this gene in ALS or FTD patients lead to mislocalization of the corresponding protein, accumulating in the cytoplasm in the form of abnormal TDP-43 aggregates and generating anomalous ubiquitin-positive inclusions in the nucleus and cytoplasm [72]. These inclusions can affect the physiological functions of p62 (also known as SQSTM), which is involved in autophagy and proteasome regulation. The sequestration of p62 within TDP-43 aggregates leads to the impairment of autophagy and proteasome functions, driving the further accumulation of misfolded proteins within cells [73,74]. Indeed, aggregates colocalizing TDP-43 with p62 and SOD1 were found in postmortem ventral spinal cord tissues of patients with fALS and sALS, despite the existence of different aggregation profiles [74]. This can occur even in the absence of mutations in the respective genes, which may be attributed to incorrect protein folding, namely SOD1 [74,75].
Another commonly mutated ALS-linked gene is FUS, which encodes the RNA-binding protein FUS. In healthy individuals, FUS is found in the nucleus and is related to gene expression regulation, DNA repair, and RNA processing [76]. However, in patients with ALS and FTD, FUS translocates into the cytoplasm, creating FUS inclusions that can boost further nefarious effects, such as RNA mislocation associated with sequestering of the motor protein kinesin-1 [77] and axonal transport impairments [78]. FUS mutations in ALS may also impair mitochondrial functions through the sequestration of respiratory chain complex mRNAs in the cytoplasm [79]. Moreover, FUS loss of function can lead to neuronal dysfunction and death [80]. It is possible that FUS mislocation into the cytoplasm may contribute to their incorporation into EVs, and then, by dissemination to other cells via intercellular transfer, the phenotype is spread into the circulation [81,82].
2. Extracellular Vesicles and Their Role in ALS Onset and Development
2.1. EVs Overview
Extracellular vesicles (EVs) are endogenous bilipid layers, plasma membranes, or endosome-derived nanoparticles released by most eukaryotic cells into the extracellular space [83]. They were first described by [84] and were initially thought to be cellular waste products. Most studies have reported that cells can synthesize and secrete three main types of EVs: exosomes (exosome-like vesicles), microvesicles or ectosomes, and apoptotic bodies [85,86]. However, more recently, other types of EVs, such as retrovirus-like vesicles and vesicles, have been reported. The former are 90–100 nm particles that possess a subset of retroviral proteins and carry endogenous retroviral sequences but not for cellular entry or retroviral propagation [87]. Mitovesicles are of mitochondrial origin, possessing components of this organelle such as mitochondrial proteins, lipids, and mitochondrial DNA (mtDNA) [88]. Mitovesicles are distinguishable from exosomes and microvesicles based on their morphology, size, and content [88]. EV classification relies on several parameters, such as size, content, function, biogenesis, and release pathways [85]. The biological functions of EVs depend on their type and highly specific bioactive cargo, which represent the progenitor cell state [89,90]. There are different ways to identify EVs, such as physical characterization through microscopy, proteomic analysis, RNA sequencing, functional characterization, and biochemical analysis of their composition [91]. An important way to identify EVs is through the presence of specific surface protein markers, which may depend on many factors, such as their origin. In the case of exosomes, some proteins tendentially common among them and often used in the identification of exosomes include annexin, CD9, CD63, CD81, HSP70, and flotillin [92,93].
It has been recognized that EVs play a fundamental role in intercellular communication, functioning as vehicles for transporting and delivering a range of cellular bioactive cargos, including membrane and cytosolic proteins, lipids, DNA, mRNA (messenger RNA), and miRNA (microRNA) [94,95]. EVs may directly influence the cellular state of recipient cells through their specific shuttled contents. This occurs via miRNA-induced gene expression posttranscriptional regulation [96], which includes numerous cellular epigenetic regulations [97,98]. EVs play a role in the maintenance of cellular homeostasis by being pivotal to cellular uptake mechanisms [99]. An example of this is the ligand/receptor interaction within brain synaptic transmission [100,101]. EVs are also important in the maintenance of stem cell plasticity [102] and in the formation of new tissues, as they are important for angiogenesis [103,104], the generation of morphologic gradients for tissue genesis during neuronal development [105,106], and neuronal regeneration [107,108].
Regarding their release pathways, EVs are delivered into the extracellular space via the SNARE-mediated fusion of multivesicular endosomes with the plasma membrane [109]. The direct budding of vesicles with the plasma membrane results in microvesicles [110,111]. Additionally, vesicles that may be shed from cells undergoing programmed cell death originate from apoptotic bodies [112]. Following exocytosis, EVs may remain in the extracellular space surrounding the secreter cell or, instead, travel elsewhere, such as in the brain, by crossing the blood–brain barrier (BBB) [113], or from the brain into the periphery. Within the brain, exosomes are released by several cell types, including neurons [114], microglial cells [115], astrocytes [116], and oligodendrocytes [117].
Different EVs are noticeable in plasma [118], urine [119], breast milk [120], cerebrospinal fluid [121], semen, peritoneal and bronchoalveolar lavage fluids, amniotic fluid, and even tumor effusions [83,86], thus allowing long-distance intercellular information exchange [89].
2.2. The Role of EVs in ALS
EVs have been associated with numerous pathologies, from metastatic cancers [122] to neurodegenerative diseases such as Alzheimer’s disease and Parkinson’s disease [123,124,125]. Under such pathological conditions, EVs shuttle enclosed misfolded proteins and other neurotoxic elements that can potentially induce dysfunction in recipient cells [125,126]. EVs are increasingly recognized as being of great importance in the pathogenesis of ALS and in the identification of biomarkers, which will be explored in this section (Figure 1).
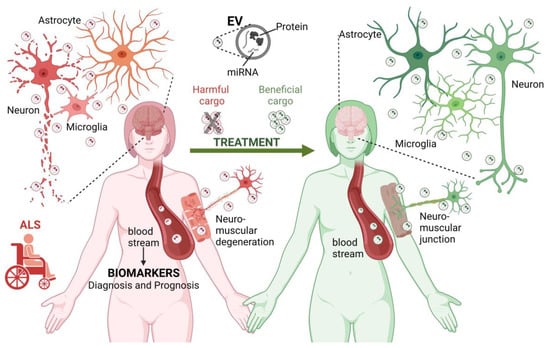
Figure 1. The role of EVs in ALS. EVs contribute to the pathogenesis of ALS (left reddish side). EVs are produced by different cell types in the central nervous system and neuromuscular junctions. In the context of ALS, EVs may carry disease-related biological molecules (proteins and miRNAs) involved in the transformation and degeneration of the brain and neuromuscular elements, thus contributing to the spread of the pathology between different cell types. Furthermore, EVs can reach long distances in the body, contributing to the exchange of harmful molecules between the brain and neuromuscular junction. Considering this, the molecules transported by EVs circulating in the bloodstream and cerebrospinal fluid are considered potential biomarkers for the diagnosis and prognosis of ALS. Finally, EVs have a therapeutic potential. Blocking the exchange of EVs carrying harmful molecules and administering EVs with neuroprotective cargo may slow the progression of ALS or revert its pathological effects (right-greenish side). ALS, amyotrophic lateral sclerosis, EV, extracellular vesicle; miRNA, microRNA. Figure created using BioRender.com (accessed on 6 June 2023).
2.2.1. EVs in ALS Disease Progression and Pathological Mechanisms
EVs have emerged as significant players in ALS progression, with increasing evidence pointing to their role in the dissemination of detrimental biocargo. EVs allow for the hypothetical prion-like propagation of ALS-related mutant misfolded proteins and dysregulated miRNAs [81], which are believed to contribute to disease severity and progression [127,128,129]. The most common cargos found in EVs from patients with ALS include misfolded proteins such as mSOD1, FUS, TDP43, and C9orf72 expansions DPRs and other neurotoxic elements [64,81]. These harmful cargos have been screened in both astrocytes and neuron-derived exosomes in different ALS disease models, such as the SOD1-G93A mouse model, which is one of the most commonly used animal models for studying ALS. In this model, the mutated SOD1 gene harbors a glycine-to-alanine substitution at codon 93. Recently, ref. [130] demonstrated that mutant SOD1 (mSOD1) accumulation occurs in cellular vacuoles, which may be constituted by different portions of organelles, and once released, leads to the existence of different types of EVs, particularly mitoEVs. The formation and type of vacuoles and the resulting EVs appear to be related to the stage of ALS pathology in this mouse model. Interestingly, before the onset of motor symptoms, these vacuoles are already present and are mainly of mitochondrial origin, with a high content of mSOD1, ultimately resulting in the release of mSOD1-containing EVs [130]. The authors of this study hypothesized that these EVs, derived from damaged neurons, may be responsible for the initiation of a sequence of signaling cascades that contribute to neuroinflammation, glial-mediated neurotoxicity, and prion-like spreading of the disease. The muscle-specific expression of mutant SOD1(G93A) can also have a negative effect on neurons, as it has been reported to alter and dismantle neuromuscular junctions through a PKCθ-dependent mechanism [131]. The expression of mutant SOD1 induces the upregulation of PKCθ, and its colocalization with acetylcholine receptors (AChR) leads to a decrease in mitochondrial function, alterations in redox signaling, and neuromuscular junctions hindering transmission [131]. Moreover, and as noted in Peggion et al. [132], myocytes from hSOD1(G93A) mice are susceptible to reactive oxygen species that lead to an unbalanced mitochondrial redox state and changes in Ca2+ homeostasis. This ultimately triggers a reactive glial response and the release of proinflammatory cytokines that affect both motor neurons and neuromuscular junctions (NMJs). To support this finding, cytokines such as IL-1b, IFN-y, and IL-6 were found in circulating EVs of the spinal cord from SOD1(G93A) mice [133]. The existence of different vacuole/EV phenotypes and associated cell death pathways may have different roles in the onset and severity of symptoms, as well as in the heterogeneity and progression of the disease.
Exosomal TDP-43 is another significant cargo that plays a crucial role in ALS progression. A longitudinal study conducted on ALS patients demonstrated an increase in the exosomal TDP-43 ratio in peripheral blood during the course of the disease, particularly in the early stages [134]. This increase in the TDP-43 ratio is associated with elevated levels of neurofilament light chain (NFL) in the plasma of these patients, which is more prevalent in individuals with rapid disease progression [134]. Further evidence supports the significance of exosomal TDP-43 in the disease propagation. Ding et al. (2015) described the damaging effect of exosomes enclosing TDP-43 C-terminal fragments (CTFs) from the cerebrospinal fluid of ALS patients with FTD (ALS-FTD-CSF) in human glioma cells (U251 cells). After incubation with ALS-FTD-CSF-derived exosomes, naive U251 cells developed intracellular TDP-43 aggregates in the form of tunneling nanotube (TNTs)-like structures [135]. Although in vivo studies are required, this previous work suggests that EVs may act as vehicles for the spread of TDP-43 aggregates in the context of ALS.
EVs and their toxic payloads not only damage neurons but also spread pathological signaling by transferring them between different cell types, including neurons, astrocytes, and muscle cells. Evidence of these interactions was provided by a study showing that the EV-mediated transfer of DRPs occurred between MNs-like NSC34 cells and rat cortical neurons and, then, from those to rat cortical astrocytes [64]. This transfer is relevant to ALS, as EVs carrying C9orf72-encoded DPRs were identified to be involved in the exchange between human C9orf72-induced pluripotent stem cell-derived motor neurons (hiPSC-MNs) and control iPSC-derived spinal MNs [64]. In NSC34 cells transfected with mutant SOD1(G93A) (hSOD1-G93A NSC34 cells), miR-124 was found to be upregulated and transferred to EVs. When these cells were cocultured with N9-microglial cells, miR-124 contained in mSOD1 exosomes was translocated to N9-microglial cells, resulting in phenotypic alterations, such as a reduction in their phagocytic capability and activation of neuroinflammation pathways [136]. Exosomes released by mouse astrocytes overexpressing G93A SOD1 were also previously shown to be responsible for the transfer of mutant SOD1 to mouse spinal neurons and to induce MN death [137]. Moreover, astrocytic-derived exosomes from the plasma of sALS patients were found to transport inflammation-related cargo, including IL-6, a proinflammatory interleukin, which was increased in these vesicles and positively associated with the rate of disease progression [138]. The negative impact of EVs and their cargo on the interaction of affected muscle cells with MNs was further demonstrated by evidence that multivesicular bodies released from ALS muscle cells are neurotoxic to healthy MNs [139]. In this study, EVs derived from muscle cells obtained from biopsies of sALS patients were exposed to healthy hiPSC-MNs and were shown to be neurotoxic through increased FUS expression, resulting in shorter and less branched neurites, atrophic myotubes, and enhanced cell death [139]. The observed cell death was greatly reduced by immunoblocking the vesicle uptake by MNs with anti-CD63. Finally, a study by Anakor et al. supported the cause and effect relationship between muscle cell vesicles and MNs. The exposure of MNs to skeletal muscle cell-derived exosome-like vesicles (MuVs) in ALS patients resulted in reduced neurite length, number of neurite branches, and reduced MN survival and myotubes by 31% and 18%, respectively. Moreover, adding ALS-derived MuVs to healthy astrocytes led to an increase in the proportion of stellate astrocytes and, thus, mild activation of these cells [140].
2.2.2. miRNAs and Misfolded Proteins EVs Cargo in ALS: Potential ALS Biomarkers
One particular cargo of EV miRNAs has attracted research interest as potential biomarkers for ALS due to their versatile functions in regulating gene expression across a wide range of processes, including neural development, cell proliferation and differentiation, protein ubiquitination, apoptosis, and other transcriptional regulatory processes. Despite their link to ALS progression, the mechanisms underlying the alterations in their expression and levels remain inconclusive. Defective RNA metabolism and miRNA dysregulation are closely associated with ALS [141]. miRNA profiles in ALS exhibit significant variations among patients and can be over- or under-expressed as they are transported by EVs across multiple biofluids and tissues. Most of the research has focused on screening plasma circulating EVs of ALS patients using a variety of research methodologies, ranging from RT-qPCR analysis to microarrays [142].
In the quest for an ALS molecular biomarker fingerprint, ref. [158] reported the downregulation of miR-27a-3p in serum-derived exosomes from patients with ALS. Saucier and colleagues [145] found 27 differentially expressed miRNAs, 5 of which were upregulated and 22 downregulated when compared via next-generation sequencing, the EVs isolated from plasma samples of ALS patients compared to those from healthy controls. Some miRNAs were relevant to ALS diagnosis, as they were related to the Revised ALS Functional Rating Scale (ALSFRS-R) scores. This was the case for miR-193a-5p, which allowed us to distinguish between patients with low and high scores. miR-15a-5p has been shown to be important in differentiating controls from patients with ALS. In a separate study, Katsu and coworkers [153] analyzed miRNA profiles in neuron-derived EVs from plasma samples of ALS patients via microarrays and identified 30 differentially expressed miRNAs: 13 upregulated and 17 downregulated. In another study, Pregnolato et al. [272] performed miRNA screening of serum-derived exosomes using RT-qPCR analysis. Owing to the small sample size used in this study, no statistically significant differences were observed in the expression levels of any miRNA. However, a recent study by Lo et al. [155] analyzed the miRNA cargo profiles of EVs isolated from postmortem homogenates of the frontal cortex, spinal cord, and serum of patients with sALS. The authors found no difference in the number of EVs between patients and controls, but patients with ALS presented larger spinal cord vesicles and smaller-sized vesicles in the serum. Two miRNAs related to axon guidance and long-term potentiation were significantly dysregulated in all analyzed tissues: miR-342-3p was upregulated, and miR-1254 was downregulated. Furthermore, the miRNA levels were reduced in the frontal cortex and spinal cord of sALS patients, whereas they were increased in the serum. Another study, performed by Rizzuti et al. [162], analyzed EVs isolated from MN cultures obtained from fibroblast-reprogrammed iPSCs of ALS patients carrying C9ORFf72, SOD1, and TARDBP mutations. These authors found the dysregulation of several miRNAs, specifically the upregulation of miR-629-5p and miR-194-5p and downregulation of miR-34a-5p, miR-1267, and miR-625-3p. Interestingly, the latter was found to be consistently downregulated in C9orf72 MN exosomes and upregulated in EVs from TARDBP-MNs. In the same study, miR-625-3p was also predicted to mediate cell-to-cell communication, immune system pathways, and autophagy. Furthermore, in another study by the same authors [273] using iPSC-derived MNs progenitors from fALS and sALS patients, further dysregulation was found, notably of miR-34a, which is involved in cell cycle regulation, autophagy, apoptosis, neurogenesis, and neuronal differentiation [274]. Sproviero et al. (2021) also searched for potential ALS EV miRNA biomarkers and found the dysregulation of hsa-miR-206, hsa-miR-205-5p, miR-1-3p, hsa-miR-205-5p, hsa-miR-200b-3p, hsa-miR-200c-3p, hsa-miR-6888-3p, hsa-miR-31-5p, hsa-miR-141-3p, and hsa-miR-210-3p in the plasma of ALS patients [275]. Using an in situ hybridization analysis, Yelick et al. (2020) found the downregulation of miR-124-3p in exosomes from SOD1-G93A mice spinal MNs. Moreover, in this study, we found a significant correlation between cerebrospinal fluid (CSF) exosomal miR-124-3p expression levels and the disease stage of male ALS patients, as denoted by the ALSFRS-R score [149]. It is worth noting that miR-124-3p is a recognized oncogene [276,277] with an essential role in cell proliferation and apoptosis [276] and is associated with poor survival rates in patients with hepatocellular carcinoma [278]. Conversely, its upregulation was shown to decrease the metastatic behavior of hepatocarcinoma cells through the reversion of CRKL expression, which resulted in the suppression of the extracellular signal-regulated kinase (ERK) pathway and inhibition of malignant cell proliferation [279]. Importantly, its upregulation was found to be protective against post-traumatic neurodegeneration through activation of the Rela/ApoE signaling pathway [280], and its downregulation was linked to the neurodegeneration and neuroinflammatory states of post-traumatic brain injuries (TBI) [281].
Other miRNAs that were differentially expressed in serum-derived extracellular vesicles from 50 patients with ALS were reported recently by [150]. Statistically significant robust results yielded a differential expression of seven miRNAs included in extracellular vesicles, two of which were upregulated (miR-151a-5p and miR-146a-5p) and three downregulated (miR-4454, miR-10b-5p, and miR-29b-3p) [150]. Among the reported functions, these specific miRNAs have been associated with tumorigenesis [282,283,284] and protection against cell apoptosis [285].
This entry is adapted from the peer-reviewed paper 10.3390/cells12131763
This entry is offline, you can click here to edit this entry!