Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Veterinary Sciences
Modern day broilers have a great genetic potential to gain heavy bodyweights with a huge metabolic demand prior to their fully mature ages. Moreover, this made the broilers prone to opportunistic pathogens which may enter the locomotory organs under stress causing bacterial chondronecrosis and osteomyelitis (BCO).
- broiler
- lameness
- bacterial chondronecrosis and osteomyelitis (BCO)
- biomarkers
- mesenchymal cells
1. Introduction
An active tissue that experiences continual remodeling is the bone. Any changes in its regular turnover may lead to skeletal diseases characterized by bone loss [1]. During the past few decades, commercial poultry production increased dramatically in terms of feed efficiency and growth rate; however, this pattern also showed some adverse implications, such as fatty liver syndrome, pulmonary hypertension, and skeletal problems [2,3,4,5]. Furthermore, modern broilers have the genetic potential to achieve higher body weights with high metabolic demands. This makes them prone to skeletal damage followed by opportunistic bacterial infection and later with bacterial chondronecrosis and osteomyelitis (BCO). The physiology of the bone and its turnover process is complex and involves several pathways working coherently [6]. A common skeletal disease condition that affects broilers worldwide is lameness and is associated with the factors including genetic traits, infectious agents, the center of gravity of the bird, body conformation, activity, and nutrition [7,8,9]. Furthermore, the rate of culling at farm level due to lameness is 0.5% to 4%, which reflects the losses of approximately $100 million per year in United States alone [9,10]. Moreover, BCO is considered as a common cause of lameness in Australia, Canada, Europe, and the US [7,11,12,13,14]. Additionally, BCO in broiler production would impact the poultry revenue through the culling and condemnation rates along with a lowered livability since the production is usually expressed as production costs, net profit per pound of a bird [15,16]. The above impact would be considerably contributed to by the incidence of lame birds in the farm, which causes higher feed conversion, lower weight gain, and higher condemnation rates in processing plant [17].
2. Inflammation and BCO
BCO is commonly observed in femurs, tibia, and thoracic vertebrae [10,18]. This occurs majorly due to formation of microfractures and clefts because of rapid growth of juvenile bones. Additionally, this is often associated with rapid increases in body weight, leading to focal ischemia providing a convenient breeding ground for bacterial colonization [18,19]. Post infection or injury, acute phase response (APR) is a key sequela that affects the nutrient requirements and metabolism, and it is usually initiated by local inflammatory response [20]. The above response can be measured by observing the changes in acute phase proteins and cytokine profiles where the excessive levels indicates decreasing production traits and rise in pathology [20,21,22,23,24]. Cytokines (IL-1,6 and tumor necrosis factor) released as a result of APR hasten bone resorption [25,26,27] as well as muscle breakdown [28]. A study involving the injection of lipopolysaccharide (LPS) to the birds showed a severe disruption in bone homeostasis, production parameters including livability, bodyweight, and feed conversion [27]. This usually occurs due to macrophages and osteoclast like cells responding to LPS by release of cytokines and nitric oxide [29]. Moreover, a study on RAW 264.7 cells in vitro were not able to differentiate into mature osteoclasts in the presence of LPS. RANKL or LPS-treated cells increased Toll-like receptor 4 (TLR4) levels in membrane [30]. A TLR4 inhibitor, TAK-242 (resatorvid) reduced the osteoclast number as well as tumor necrosis factor (TNF)-α in LPS treated cells. In contrast, RANKL- induced cells were not affected by TAK-242 and secreted basal levels of TNF-α. This clearly shows that LPS associated bone resorption is associated with LPS/TLR4/tumor necrosis factor receptor (TNFR)-2 axis but not with RANKL/RANK/OPG axis [30]. Furthermore, osteoclasts and their activities are regulated by osteoblasts which, in turn, are altered by the bacteria and their products by means of apoptosis which occurs by activation of intrinsic and extrinsic cell death pathways leading to disruption in bone homeostasis [31] (Figure 1).
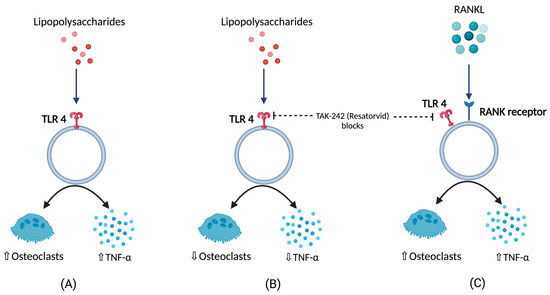
Figure 1. (A) Response of lipopolysaccharides (LPS) with respect to TNFα (tumor necrosis factor), and osteoclastic activity (B) TNFα, and osteoclastic activity in the presence LPS under the influence of TLR4 blockers (C) TNFα, and osteoclastic activity in the presence of RANKL under the influence of TLR4 blockers.
3. Pathogens and BCO
Staphylococcus aureus is the most commonly reported organism found in BCO epiphyseal lesion, but septicemic pathogens like Staphylococcus hyicus, S. xylosus, S. simulans, Mycocaterium avium, Salmonella spp., E. coli, and Enterococcus were also isolated [10,13,32,33]. Histological changes in epiphyseal region of growth plate due to BCO lead to transection of the capillaries and blood vessels within the highly vascularized epiphyseal region, which paves the way for decreased blood flow to the areas around that region [18]. Furthermore, this allows the circulating bacteria to enter and proliferate, along with immunological response leading to tissue damage (necrotic abscesses and voids) [14,18,19,34]. Physiological stress also aids in heightened entry of opportunistic pathogens through the epithelial tight junctions, and the pathogens finally arrive at osteochondrotic micro-fractures and clefts [35]. BCO is also observed in clinically non-lame birds at higher incidences, but there are notable pathognomonic lesions seen in lame birds with BCO [14,34].
Bacterial translocation through the damaged epithelium is reported to be one of the causes for higher incidence of BCO [9]. In addition, young intestines have higher susceptibility to bacterial leakage than fully developed intestines in the presence of mucosal damage [9,34]. Furthermore, chicken anemia virus and infectious bursal disease virus make the birds more prone to BCO as a result of immunosuppression [10,36]. An understanding of bacterial diversity, 3D structural alterations in bone and cartilage, bone remodeling marker gene expression, and omega 3 fatty acid and/or probiotic supplementation in BCO is still an important topic that is focused on; however, it is necessary to be further rationalized to identify precise etiology and treatment. Additionally, there is a limited information on extra intestinal bacteria which may induce the APR followed by BCO. In a study involving linear discriminant analysis effect size (LEfSe), an analysis showed that physiological stress will allow commensal and pathogenic bacteria to enter extra intestinal sites, indicating the need for exploring the novel taxa [37,38]. These extra intestinal sites would be circulating maternal blood microbiota in chick or in vivo colonies of microbes [37]. Apart from microbiota, some managemental practices like light intensity, drinking water, and flooring may also affect the incidence of BCO [39]. Light intensity is directly related to the bird movement and activity which may affect the incidence of BCO [40,41,42,43], and a study showed that detecting surface temperatures of broiler leg regions with the help of non-invasive methods would also help in detecting lesions of BCO [44]. In two different studies, provision of 25-OH vitamin D3, prophylactic administration of probiotics were reported to abate the incidence of BCO in wire flooring model which were attributed to trigger the lameness in broilers [34,45] (Figure 2).
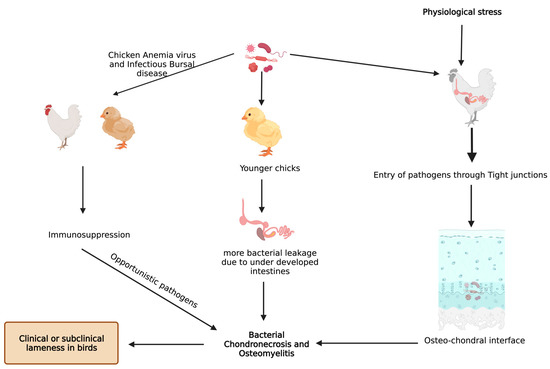
Figure 2. Known pathways through which bacterial chondronecrosis and osteomyelitis (BCO) is observed in modern day broilers.
4. Underlying Anatomy and Physiology behind BCO and Its Potential Biomarkers
BCO and other bone problems usually occur in the birds with higher growth rate and body gain which may be related to different breeds of commercial poultry [46,47]. Mostly, the body gain is attributed to pectoral muscles resulting in a shift in center of gravity with a disproportionate development of femur, which makes them more prone to BCO [48,49,50,51,52]. Integrity of articular cartilage (AC) and growth plate cartilages are crucial because the disproportionate development of legs to the body predisposes to injury under strenuous conditions. These cartilages greatly differ in their histology and extracellular matrices. AC is made of mostly all collagens except type X collagen and proteoglycans with chondrocytes [53,54]. Type X collagen appears in the growth plate usually when it undergoes the endochondral ossification process, which is assisted by adhesion molecules like cadherins and integrins, essential for regulating canonical signaling in Wnt pathway, which is activated upon binding of Wnt ligands to LRP-5/6 co receptors, and this can be inhibited when these receptors are bound to Wnt antagonists like sclerostin and Dkk-1 (Dickkopf proteins) [55,56,57](Figure 3). Among these adhesion molecules, cadherins mediate homotypic adhesion between bone cells, and integrins mediate adhesion between bone cells and its extracellular matrix [58]. Usually, long bones will develop by endochondral ossification where mesenchymal stem cells forms chondrogenic template through chondrogenic differentiation followed by hypertrophic differentiation, resulting in blood supply and remodeling of chondrogenic template into bone through the release of angiogenic factors [59]. Furthermore, the gradient increase in oxygen levels is also important in modulating the endochondral ossification where chondrogenic differentiation takes place at low levels of oxygen and hypertrophic differentiation at higher levels of oxygen tension [60,61]. The proximal tibial center is the only true secondary ossification center in the long bones of fowl during the rapid growth phase, leading to a reduced reinforcement of AC [62,63]. Although there is no conclusive evidence, ischemia is presumed to be the cause of lowered blood supply, leading to poor reinforcement of AC [19].
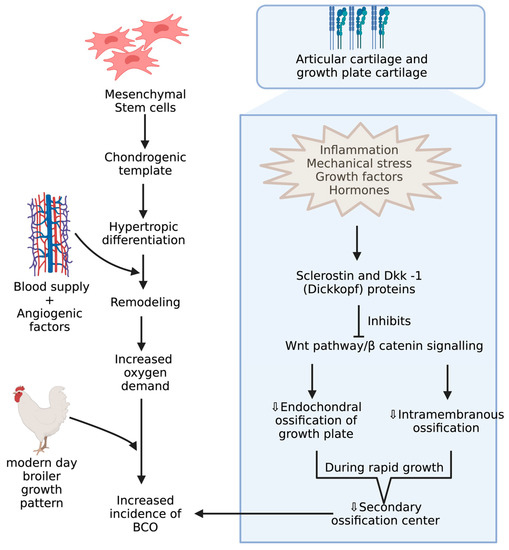
Figure 3. Mechanisms during pre and postnatal development of chicken increasing incidence of BCO.
Osteochondrotic crypts are developed from poor mineralization of chondrocytes, resulting in microfractures that allow the opportunistic bacteria to colonize in those crypts through hematogenous routes [18,19,35,64,65,66,67]. These bacteria may have originated from broiler breeders, hatchery contamination, a gastrointestinal tract, a respiratory system, or an integumentary system [19,68,69]. In an experimentally induced and spontaneous occurring study on BCO in broilers, dyslipidemia was reported to be a common feature [70]. Moreover, thrombospondin, interferon γ, and transforming growth factor-ß, and angiogenesis inhibitors was suggested to be the risk of avascular necrosis [71,72]. In young chickens, the arrest of angiogenesis and growth plate development due to reduction in plasma levels of vascular endothelial growth factor isoform-C and protochaderin-15 (adhesion molecule) occurs in glucocorticoid-induced BCO, leading to apoptosis of chondrocytes [3,73]. Furthermore, reduced levels of fibroblast growth factor-2 and runt related transcription factor-2 (RUNX2), which are regulators of apoptosis and chondrocyte maturation, respectively, were suggestive of promoting BCO [74,75]. In some studies, serum metabolites like lipids, lipoproteins, and apolipoprotein-derived peptides showed changes when chickens were induced with glucocorticoids and naturally occurring BCO [3,70,76]. On the other hand, the use of these biomarkers for BCO needs further validation because they were significant in vascular diseases and osteoarthritis [77,78].
An experimental model of chicken BCO for human osteomyelitis identified a novel pathophysiological mechanism for this severe inflammatory condition, which is described below [79]. DICER1 (a highly conserved RNaseIII endoribonuclease), a multifaceted protein which is responsible for dsRNA cleavage and its dysregulation, was recognized in several human diseases and was reported to have a critical role in osteogenesis [80,81,82,83,84,85]. DICER1 dysregulation alters cortical bone integrity and homeostasis which are usually associated with RUNX2 [86]. DICER1 dysregulation and infection exposure leads to an increase in dsRNA levels [79]. A study showed DICER1 dysregulation due to bacterial infection might induce dsRNA accumulation, which, in turn, is related to the IL-1β pathway, which plays a key role in pathogenesis of human bone inflammation. DICER1 dysmetabolism acts as an upstream regulator of NACHT (nucleotide-binding domain, LRR (leucine-rich repeat), and PYD (pyrin domain) domains containing protein (NLRP)3 inflammasome, upon activation of NLRP3, and paves the way for a break in bone homeostasis through the increased activity of neutrophils, monocytes, macrophages, osteoblasts, and osteoclasts [87,88,89,90,91] (Figure 4). Although there is a potential positive impact from inflammasome activation through a reduction in bacterial proliferation and removal of a pathogen from the host, this decreases the osteoblastic activity. Additionally, NLRP3 levels tend to be higher in bone tissue affected by a pathogen than the one with fractures, indicating that this inflammasome activation contributes to inflammatory bone loss [79,91]. MtDNA mutations are one form of mitochondrial dysfunction associated with alterations in mitochondrial biology [92,93]. This is usually associated with Alzheimer’s disease, dementia, coronary heart disease, chronic fatigue syndrome, and ataxia [92,93,94,95,96,97,98]. This association with several diseases is due to the relation between its dysfunction to apoptotic and inflammatory pathways. Mitochondria are direct targets for some bacterial infections like Staphylococcus aureus [96,99].
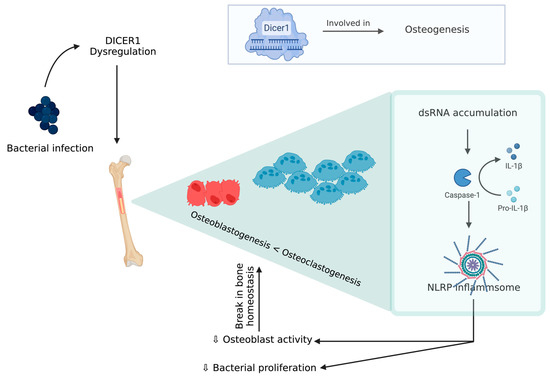
Figure 4. DICER 1 dysregulation in the presence of bacteria causing break in bone homeostasis.
Peroxisome proliferator-activated receptor coactivator-1 (PGC-1α and PGC-1β) targets transcription factors (transcription factor A mitochondrial) and gene expression in mitochondrial biogenesis pathways [100]. Precisely, any changes in metabolism or cell growth will modulate the expression and upregulation of PGC-1α, leading to an increased mitochondrial biogenesis and respiration in inflammatory states [101,102]. Both mitochondrial biogenesis-associated genes (PGC-1α and PGC-1β) are significantly upregulated in BCO-affected tissue. In addition, the inflammatory response, associated reactive oxygen species accumulation, and metabolic shifts cause an increased need for mitochondria. On the other hand, during stress, mitochondrial fusion occurs, leading to formation of a large network, and this is associated with important components such as OPA1 (mitochondrial dynamin like GTPase) and Mitofusins (MFN1 and 2) [103,104]. Additionally, in ascites, OPA1 expression is decreased in the susceptible lines but not in ascites-resistant selected line [105]. In contrast, BCO showed a significant decrease in OPA1 but an upregulated MFN2 [106]. The decrease in the former is coupled with a gradual increase in OMA1 that is a regulator of mitochondrial fission via cutting OPA1 at some sites and making it inactive. At high levels of cellular stress, fission causes removal of damaged mitochondria when complementation through fusion is not possible. The above shift from fusion to fission indicates mitochondrial turn-over in accordance to the high level of stress during BCO [106]. Additionally, other potential and widely studied biomarkers are shown in Table 1.
Table 1. Represents the potential biomarkers involved in the incidence of BCO.
| Biomarkers | Function and Correlation | References |
|---|---|---|
| Serum Calcium | Bone density and mineralization along with bone breaking strength | [107,108] |
| IL-17, IL-6, TNF-α, NLRP-3 | Pyroptosis of osteoblasts, and Pro-inflammatory factors stimulates osteoclastogenesis or inhibits osteobalstogenesis | [106,109] |
| Peroxisome proliferator actvated receptor coactivator (PGC-1α, 1β) | Repress the transcriptional activity of NF-κB, Mitochondrial biogenesis | [106,110,111] |
| Mitofusins | Increased ROS production, | [106] |
| Matrix metalloproteases | Tissue remodeling, angiogenesis, Extracellular matrix degradation | [107,112] |
| Osteocalcin | Secreted by differentiating osteoblasts | [14,75,113] |
| RANKL and OPG | Crtitical cytokine produced by osteoblasts and OPG is an decoy receptor for RANKL | [83,114,115] |
| Alkaline phosphatase | Involved in Ca and P deposition during the bone mineralization and formation | [9,111,116] |
| Sclerostin, DICKKOPF protein | Inhibit Wnt/β-catenin signalling pathway | [117,118,119,120] |
| Tartarate resistant Acid Phosphatase | Activity of osteoclasts | [19,109,111] |
| Thrombospondin, Interferon-γ, Tranforming growth factor-β, Vascular endothelial growth factor isoform-C, and Protocadherin-15 | Associated with the risk of avascular necrosis seen in BCO | [9,19,69,121] |
| Fibroblast growth factor-2, BMP, SMAD1 and RUNX-2 | Essential in osteoblast activity, bone mineralization, and osteoclast differentiation | [4,32,122] |
This entry is adapted from the peer-reviewed paper 10.3390/biom13071032
This entry is offline, you can click here to edit this entry!