For many years, scientists have faced challenges in solving biological problems with precision and efficiency. Therefore, precision biology has emerged as a field that provides more efficient and accurate means for life science research [
1,
2]. Precision biology is a discipline focused on elucidating the biological effects of genes, proteins, and other biomolecules through the analysis of their intricate interactions. It has evolved from the field of precision medicine and extends beyond the realm of human biology. Precision biology investigates the molecular pathways that underlie disease onset and progression by considering individual variations in genes, environment, and lifestyle factors [
3,
4,
5]. It can help researchers to identify specific targets for the development of drugs and the design of personalized treatment regimens [
6,
7,
8]. Complementing precision biology, recent advances in three-dimensional (3D) genomics enable researchers to reveal the overall pattern of transcriptional regulation of genes on a genome-wide scale [
9,
10]. This clarification helps identify the regulatory elements required to transcribe specific genes and understand how they interact with other genes [
9,
10].
Transcriptional regulation, as a determinant in gene expression, is accompanied by a large number of changes in regulatory elements and is particularly important in precision biology [
11]. Genes exhibit different transcriptional patterns, such as expression or shutdown, when interacting with different transcriptional elements or protein molecules. The gene expression level also increases or decreases continuously with changes in the concentration of transcriptional elements or protein molecules in the gene expression pattern [
12]. 3D genomics reveals the interactions between different genes and transcriptional elements in cells and even organisms, which can be used to determine the specific spatiotemporal expression patterns of genes that may depend on these interactions [
12]. These minor changes in the expression pattern of intranuclear target genes caused by different interactions of genes or transcription elements can be directly used in the study of precision biology and will directly advance the development of precision biology. In this context, special attention is given to enhancers, which are regulatory elements involved in enhancer-mediated transcriptional regulation.
There is a large number of non-coding DNA and RNA regulatory elements in the genome, and enhancers are one type of non-coding DNA sequences [
13,
14]. They are mainly found in intergenic and intronic regions and activate the expression of target genes [
15,
16]. As cis-regulatory DNA elements in eukaryotes, enhancers bind tissue-specific transcription factors (TFs) and specifically transmit regulatory information to their target gene promoters, participating in transcriptional regulation [
17,
18]. Through continuous research and accumulated datasets, scientists have realized that the number of enhancers is far greater than the number of genes [
19,
20,
21]. Enhancer-mediated transcriptional regulation is tightly correlated with spatiotemporal gene expression patterns. Enhancers are crucial for the proper development and function of organisms, as they determine the specificity of cellular responses to environmental stimuli and regulate biological processes at the molecular level [
22]. However, there is still a lack of studies on the regulation of the same gene by multiple enhancers at different stages of growth and development. It is essential to resolve the patterns of multi-enhancer regulation. In recent years, super enhancers have also received more attention due to their key role in controlling cellular identity and disease, especially cancer [
23,
24,
25]. Super enhancers are clusters of enhancers that have more complex mechanisms of action, providing a more appropriate explanation for fine transcriptional regulation. In other words, researchers can start with enhancers to explore the three-dimensional spatial conformational information and study the different functions of genes. This approach can help dissect the possible patterns of spatio-temporal specificity in gene expression within transcriptional regulator networks.
2. The Development of Precision Biology
Precision biology has emerged from precision medicine, aiming to gain a more complete and accurate understanding of biological systems through the utilization of advanced technologies and methods. The concept of precision medicine was introduced by the National Institutes of Health (NIH) in 2015, with the goal of providing personalized information to individuals and their families to improve health outcomes [
27]. Precision medicine represents a novel approach to disease prevention and management [
1,
28] that considers individual genetic, environmental, and lifestyle differences. Its primary focus is on treating cancer, while the long-term objective is to establish a new model of medicine that emphasizes individualized engagement, data sharing, and privacy protection [
29,
30]. The integration of artificial intelligence (AI) and machine learning (ML) makes it possible to employ computational approaches in precision medicine [
31,
32]. These approaches enable the identification of drugs that can be repurposed for other diseases, utilizing diverse sources of data ranging from molecular signatures to the effects of drugs at the molecular and system levels. In precision biology, advanced technologies and methods are employed, including high-throughput experimental techniques, such as genome sequencing, proteomics, metabolomics, and single-cell analysis. These techniques generate large amounts of data, which can then be analyzed using computational methods [
27,
33].
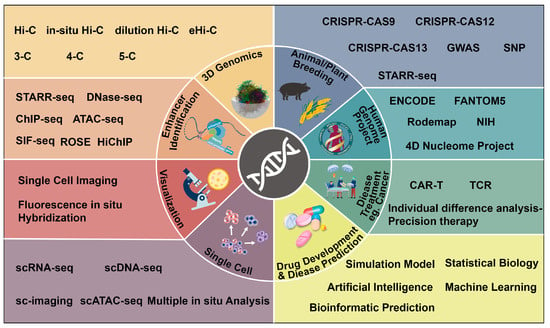
In the previous section, we mentioned that subtle changes in gene expression patterns resulting from interactions between genes and transcriptional elements (especially enhancers) play a direct role in precision biology research. To investigate these “minor changes”, a number of 3D genomics techniques have been developed. These include chromosome conformation capture (3C)-based technologies, such as chromosome conformation capture-on-chip (4C) [
34], chromosome conformation capture carbon copy (5C) [
35], high-throughput/resolution chromosome conformation capture (Hi-C) [
36], Single-cell Hi-C [
37], eHi-C [
38], in 3D genomics, chromatin interaction analysis by paired-end tag sequencing (ChIA-PET) [
39], and related methods, which have enabled the discovery of transcriptional regulatory elements, especially enhancers, and their effects on gene expression (
Figure 1). In addition, various methods, such as self-transcribing active regulatory region sequencing (STARR-seq), chromatin immunoprecipitation sequencing (ChIP-seq) [
40,
41], DNase-seq, site-specific integration FACS-sequencing (SIF-seq), assay for transposase accessible chromatin with high-throughput sequencing (ATAC-seq), robust statistical estimation (ROSE) algorithm, in situ Hi-C followed by chromatin immunoprecipitation (HiChIP), have been widely used for the identification and annotation of enhancers and super enhancers (annotations of enhancer and super enhancer are described in detail in
Section 4). These techniques significantly contribute to the development of 3D genomics.
Figure 1. List of technologies that promote the development of precision biology. The multidimensional resolution of precision biology (inner circle), the advancement of precision biology through various enabling milestones through different translational axes, such as high-throughput research methods and big data analysis and integration (outer frame).
For instance, researchers have utilized Hi-C and ChIP-seq to investigate the impact of MED23 mutations on enhancer activation and chromatin conformation. These studies have provided insights into the pathogenic mutations of MED23 and the role of mediator in transcriptional control based on enhancer topology [
42]. Single-cell Hi-C has been used to systematically map the three-dimensional genomic and dynamic epigenetic profiles of macrophages during differentiation and immune response [
43]. This approach has revealed the potential mechanisms underlying the reprogramming of gene expression profiles in immune cells (
Figure 1). The SIF-seq has been developed to evaluate enhancer activity in various disease-associated cell types. It has successfully identified cardiac enhancers in in vitro differentiated cardiomyocytes and neuronal enhancers in neural progenitor cells [
44], contributing to our understanding of disease-related mechanisms. STARR-seq has been instrumental in identifying regulatory variants associated with cancer susceptibility [
45]. It has contributed to the interpretation of genome-wide association studies (GWAS) results and provided valuable information for cancer risk assessment. Moreover, these cutting-edge technologies have also been used to uncover potential target enhancers for agronomic traits in plants, such as salt stress tolerance [
46], facilitating the achievement of precision crop breeding. These advanced techniques have significantly expanded scientists’ knowledge of the relationship between genome-wide spatial conformation and gene transcriptional regulation, thereby driving the progress of precision biology.
On the other hand, recent changes in sequencing technology have led to the rapid development of single-cell sequencing [
37], enabling researchers to gain insights into cellular information at the individual cell level and creating new opportunities for studying enhancer-mediated transcriptional regulation. Single-cell omics and imaging techniques have become essential tools for the realization of precision biology [
47,
48,
49]. These techniques, including single-cell RNA sequencing (scRNA-Seq), single-cell DNA sequencing (scDNA-Seq), and single-cell assay for transposase accessible chromatin with high-throughput sequencing (scATAC-Seq), as well as various in situ analysis methods, allow for the measurement of gene expression, mutations, epigenetics, and open chromatin regions. Such information is crucial for resolving tumor heterogeneity, characterizing the tumor microenvironment, and identifying rare cell subpopulations [
50,
51]. Furthermore, advancements in imaging techniques have facilitated the study of single-cell imaging, overcoming the limitations of single-cell histology techniques in capturing and visualizing the spatial relationships of cells within their native tissue environment [
52]. Spatially single-cell epigenomics approaches have enabled the labeling of histone modifications of active promoters, putative enhancers, and silent promoters, enabling spatial analysis of epigenetic modifications and DNA-binding proteins [
53]. These techniques have advanced our understanding of how gene expression is regulated spatially and temporally by the epigenome. By simultaneously capturing genomic output at different levels, single-cell techniques allow scientists to document cellular properties and functions [
54,
55]. Epigenomic technologies play a crucial role in understanding cellular diversity and uncovering mechanisms of gene regulation. This, in turn, contributes to the discovery of enhancer-mediated transcriptional regulation. It has been observed that there is a synergistic effect of tumorigenesis among super enhancers [
56]. Therefore, understanding the mechanism of enhancer-mediated transcriptional regulation from a single-cell perspective can provide a new level of understanding of cell fate determination, identity, and function in normal development, physiology, and disease. Careful and comprehensive analysis of individual cells within a population will likely completely change the way we describe and treat disease and may even explain some of the different responses that occur in patients with similar conditions and treatment processes.
Developments in bioinformatics and computational biology have further improved data availability and accuracy, contributing to accurate screening enhancers and enhancer-promoter interactions from large sequencing datasets [
57,
58]. Furthermore, the Human Genome Project (HGP) [
59], the Human Genome Encyclopedia (ENCODE) Project [
13], and the ongoing 4D Nucleosome Project [
60] are progressing well thanks to the infrastructure of systems biology (
Figure 1). These initiatives provide valuable resources for precisely targeting, screening, and comprehensively characterizing the enhancer-mediated transcriptional regulation network.
The advancements in precision biology have yielded remarkable progress in medical research, leading to improved healthcare standards and overall public health. In the pursuit of precision biology, to explore the individual differences and mutations that exist in individual genomes and how these differences and mutations affect the physiological and pathological characteristics of cells, tissues, and individuals, transcriptional regulation, especially enhancer-mediated transcriptional regulation, serves as a crucial tool and platform for precision biology.
3. Transcriptional Regulation and Precision Biology
Transcriptional regulation is commonly believed that occurs at two interrelated levels: The first involves transcription factors and the transcriptional apparatus, while the second involves chromatin and its regulators, including enhancers. Researchers are currently striving to elucidate how mammalian gene expression is temporally and spatially constrained by integrating these two aspects [
16,
61].
4. The Role of Different Enhancers in Transcriptional Regulation
Compared to the intricate pathways and diverse transcriptional machinery involved in parsing transcriptional regulation, analyzing the differences in gene expression attributed solely to enhancers can provide insights into the role of transcriptional regulators from a three-dimensional genomics perspective. This approach can help identify transcriptional regulators to some extent.
4.1. Typical Enhancers
Typical enhancers (TEs) are short genomic DNA sequences. They are usually considered to have the following characteristics, including: Enhancing the expression of the target gene, the function independent of position, being located in the open chromatin region, exhibiting enrichment of transcriptional co-activators with histone modifications, and containing specific DNA sequences that allow for binding TFs [
17]. The first enhancer discovered was a 72 bp-long DNA fragment from the late gene region of simian virus SV40, which increased the expression of a reporter gene promoter by ~200-fold [
92,
93].
4.2. Annotation of Typical Enhancers
To explain enhancer-mediated transcriptional regulation and understand E-P interactions, scientists need to systematically annotate enhancers. In the genomic era, various methods have been employed for enhancer annotation. For example, histone-modified ChIP combined with microarrays (ChIP-ChIP) and next-generation sequencing (ChIP-seq) to predict enhancers [
94,
95,
96]. Currently, commonly used methods for enhancer annotation include: Integration of DNase hypersensitivity analysis with deep sequencing, detecting of a higher proportion of histone H3 lysine 4 monomethylation (H3K4me1) compared to trimethylation (H3K4me3), assessing histone acetylation (e.g., H3 acetylated at lysine 27 (H3K27ac)), identifying certain histone variants (e.g., H2AZ), examining the binding of co activator to acetyltransferases (e.g., creb-binding protein with p300 (CBP/p300)), and analyzing the aggregation binding of multiple TFs [
79,
96,
97,
98]. Thousands of enhancers in various animal models, including drosophila, nematodes, mice, and humans have been annotated by different international genome annotation consortia such as ENCODE [
13,
99], NIH Epigenome Roadmap [
100], FANTOM5 [
101,
102], and Blueprint/IHEC [
103]. Additionally, enhancer-related databases such as VISTA enhancer Browser [
104], enhancer Atlas [
105], and Human Enhancer Disease Database (HEDD). [
106] have been developed to visualize and share enhancer annotation information among mammals (
Figure 1). These resources provide valuable insights into the role and mechanisms of enhancer-mediated gene regulation. The use of epigenomic markers has shifted towards identifying developmental enhancers that are more likely to drive tissue-specific patterns of gene expression [
95,
107]. However, the annotation of epigenomic features has generated a large number of putative enhancers in humans (ranging from 400,000 to 1 million), which is more than ten times the number of coding genes. Importantly, the presence of histone modifications does not fully explain the molecular mechanisms underlying enhancer activity [
95,
108]. Arbitrary truncation based on the H3K4me1/H3K4me3 ratio to select enhancers may miss some functional enhancers [
109]. These findings suggest the need for additional criteria to annotate functional enhancers more precisely in the genome.
4.3. Super Enhancer
Cells rely on 3D genomic organization for transcriptional control, and they also utilize super enhancers (SEs) to regulate transcription. SEs are a class of cis-regulatory elements with super-transcriptional activation properties. In 2013, Richard A. Young’s lab proposed the concept of SEs [
23] based on the study of enhancers at that time. Super enhancer regions typically span 8–20 Kb, which is much larger than the 200–300 bp span of normal enhancers. They are a large cluster of transcriptionally active enhancers enriched with a high density of master transcription factors (
Figure 3), cofactor, and enhancer histone modification marks that control cell identity gene expression [
110,
111]. These SEs can explain cell type-specific expression patterns [
112] and have significant potential for applications in developmental biology [
113], cancer, and other disease [
113,
114,
115] pathogenesis studies.
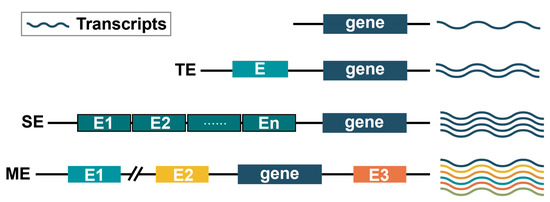
Figure 3. Comparison of typical enhancer (TE), super enhancer (SE) and multi-enhancer (ME) interoperability patterns. (1) TE usually positively drives target gene expression and more transcripts are generated. E denotes a single enhancer. (2) SE is usually a cluster of enhancers consisting of neighboring enhancers. SE has a stronger effect on gene transcription enhancement than TE. It can help to produce more transcripts. E1 to En denote different enhancers. (3) In ME modulation, enhancers may be distributed at different positions of the gene and may be far from the gene, such as E1 or be adjacent to the gene, such as E2. Each enhancer can enhance gene expression, but the resulting transcriptions may be different. Different transcriptions are shown in different colours.
4.4. Multi-Enhancer Modulation
Based on the specificity of transcriptional regulation exhibited by both TE and SE, gene regulation often involves multi-enhancers that jointly control transcription [
128,
129] and the enhancer hub mentioned above. We propose a model of multi-enhancer co-regulation, which plays a critical role in transcriptional regulation (
Figure 3). Multi-enhancers (MEs) can be located on the same side or on different sides of the gene, and they can even be positioned far away from the gene. Different enhancers may act at different sites, resulting in various phenotypic changes. Collaborative or competitive relationships may exist among multi-enhancers, including super enhancers [
56]. Therefore, understanding the molecular mechanisms of multi-enhancer regulatory patterns can provide insights into development, disease, physiology, and biology, ultimately driving advancements in precision biology.