Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Gastroenterology & Hepatology
Hepatocellular carcinoma (HCC) is the most common primary liver malignancy. The hypervascular nature of the majority of HCCs and the peculiar vascular derangement occurring during liver carcinogenesis underscore the importance of angiogenesis in the development and progression of these tumors. Indeed, several angiogenic molecular pathways have been identified as deregulated in HCC. The hypervascular nature and the peculiar vascularization of HCC, as well as deregulated angiogenic pathways, represent major therapeutic targets.
- hepatocellular carcinoma
- angiogenesis
- tyrosine kinase inhibitors
1. Introduction
Hepatocellular carcinoma (HCC) represents the sixth most commonly diagnosed cancer and the third leading cause of cancer-related death globally [1]. Approximately half of HCC patients are diagnosed at advanced tumor stages, precluding potentially curative treatments such as surgical resection or liver transplantation [2]. As a consequence, the prognosis of patients with HCC is very poor with 5-year survival of 20% [3].
Angiogenesis, one of the fundamental hallmarks of cancer [4], plays a pivotal role in the development and progression of HCC, which is typically a hypervascular tumor [5,6]. Since, the growth of liver tumor requires the formation of new blood vessels, HCC displays intense neoangiogenic activity during its development. Moreover, a peculiar vascular derangement occurs during liver carcinogenesis, since the tumor tends to be almost entirely fed by arterial inflow, unlike the surrounding parenchyma that receives the majority of blood supply through the portal system [7]. However, in liver tumors, newly formed blood vessels display marked vascular abnormalities, which may further activate angiogenic pathways, leading to a vicious cycle. It has been demonstrated that the overactivation of angiogenesis in HCC is associated with worse prognosis. A transcriptomic signature of five genes involved in the angiogenetic process (ANGPT2, NETO2, ESM1, NR4A1, and DLL4) was found to accurately identify rapidly growing tumors and was associated with shorter survival [8]. In addition, several studies suggest that overexpression of vascular endothelial growth factor (VEGF) and its transcription factor hypoxia-inducible factor (HIF)-1α, the two key mediators of angiogenesis, is a negative prognostic factor, particularly in patients treated with surgery and systemic therapies [9,10,11,12,13,14,15,16,17,18,19,20].
The very important role of angiogenesis in the development and progression of HCC provides a strong rationale for antiangiogenic strategies as therapy. Angiogenesis has always been considered an important therapeutic target in these patients. Intra-arterial locoregional treatments (IATs) (i.e., transarterial embolization (TAE) and transarterial chemoembolization (TACE)) are commonly applied treatments for HCC worldwide [21]. Their activity is completely or in part reliant on the embolization of tumor feeding arteries with the aim of achieving tumor ischemic necrosis. Considering systemic therapies, over the last decades, multiple antiangiogenic therapies have been developed. In fact, most currently approved treatments for advanced HCC in the first- and second-line settings target angiogenic pathways [22]. More recently, the combination of an immune checkpoint inhibitor (ICI) anti-programmed death ligand 1 (PD-L1) (atezolizumab) and a monoclonal antibody targeting VEGF (bevacizumab) demonstrated a clear survival benefit over sorafenib [23]. Considering that previous trials with anti-PD1 immune checkpoint inhibitors alone (nivolumab and pembrolizumab) failed to show efficacy in first- and second-line treatment [24,25], these results seem to further confirm the importance of angiogenic pathways in the progression of HCC.
2. Angiogenesis in Hepatocellular Carcinoma
Normal liver receives approximately 26% of cardiac output (~1.1 mL O2/g/min) and consumes approximately 20% of the total O2 used by the body at rest (~0.06 mL O2/g/min) [26]. About 75% of the hepatic blood supply is received by the portal vein, while the rest is received by the hepatic artery. Lobules are the fundamental unit of normal liver: they are segregated by interlobular connective tissue and contain “cords” of hepatic parenchymal cells (hepatocytes), separated by vascular sinusoids. Sinusoidal endothelium is fenestrated and lacks a basement membrane, therefore permitting blood plasma to surround hepatocytes through the space of Disse. Other cells involved in liver physiology are hepatic stellate cells, also known as pericytes, and Kupffer cells, which are resident liver macrophages. Stellate cells are closely linked to sinusoids in the space of Disse and play a crucial role in liver fibrosis after liver damage. In the hepatic sinusoids, arterial (from the hepatic artery) and venous (from the portal vein) blood mix together, and after being “filtered” by hepatocytes, this blood flows out of the lobule through the central hepatic vein.
In liver tumors, newly formed blood vessels display marked vascular abnormalities [27,28], leading to hypovascular areas and severe hypoxia and/or necrosis and causing further stimulation of angiogenesis. Although HCC is a highly angiogenic cancer, it seems to be characterized by hypoxia [5], which has been associated with HCC growth, progression and resistance to therapies [29]. Nevertheless, while some characteristics of HCC (hypervascularity, areas of necrosis and primary resistance to therapy) suggest the presence of severe hypoxia, direct evidence of hypoxia in human HCC is missing [26]. In fact, pO2 in human HCC have not yet been accurately measured directly, and thus, the relevance of hypoxia in determining hypervascularity and arterialization of HCC is still unproved [26]. Moreover, it is important to keep in mind that the activation of angiogenic pathways (HIF target genes) can be achieved by various hypoxia-independent mechanisms [30].
The destabilization of the microvasculature, leading to vascular hyperpermeability, remodeling of the extracellular matrix and endothelial cell activation, is a fundamental step for the initiation of angiogenesis [5]. Activated endothelial cells form new blood vessels by proliferating, migrating and undergoing cord formation. Subsequently, recruited and activated pericytes stabilize the newly formed blood vessels [31,32,33]. During physiological angiogenesis, the release of antiangiogenic molecules balances the expression of proangiogenic factors [34]. By contrast, as shown in Figure 1, tumor-induced angiogenesis results from an imbalance between proangiogenic factors (VEGF-A, -B, -C and -D, angiopoietins, fibroblast growth factor (FGF), hepatocyte growth factor, endoglin (CD105), platelet-derived growth factor (PDGF), and others) and anti-angiogenic molecules (angiostatin, thrombospondin-1, endostatin, and others) [22]. In the following paragraphs, the roles of the main molecules involved in angiogenesis are reviewed briefly.
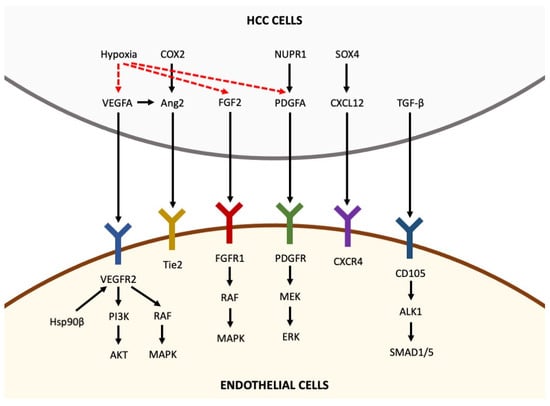
Figure 1. Pro-angiogenic factors inducing angiogenesis in hepatocellular carcinoma. Pro-angiogenic factors, including VEGFA, Ang2, FGF2, PDGFA, CXCL12 and TGF-β, are secreted by HCC cells and bind to their receptors expressed in endothelial cells, thus activating intracellular pathways that promote angiogenesis. Hypoxia is able to upregulate the expression of VEGFA, FGF2 and PDGFA in HCC cells (the arrows indicate the sequentiality of molecular pathways).
2.1. Hypoxia-Inducible Factor 1 (HIF-1)
HIF-1 is a heterodimer composed of two subunits, HIF-1α and HIF-1β. The former is an oxygen-sensitive subunit whose expression is induced under hypoxic conditions, while HIF-1β is constitutively expressed [35]. Regardless of O2 levels, HIF-1α is constitutively transcribed and synthesized through a series of events involving different growth factors and signal molecules. Under normoxic conditions, HIF-1α undergoes rapid degradation by proteasome and ubiquitination within a pathway involving the Von Hippel Lindau protein (pVHL), a tumor suppressor protein that is part of ubiquitin-ligase E3, which recognizes HIF-1α following its prolyl-hydroxylation by proteins containing protohydroxylases (PHD1, PHD2 and PHD3). In addition, asparagine hydroxylation of HIF1α blocks its interaction with the transcriptional co-activators, CREB-binding protein (CBP) and p300. By contrast, under hypoxic conditions, several pathways control the stability and transcriptional activity of this subunit through post-transcriptional modifications, including hydroxylation, acetylation, ubiquitination and phosphorylation reactions [36,37]. Being oxygen-requiring processes, neither hydroxylation nor acetylation of proline and lysine residues can occur under hypoxic conditions, resulting in greater stability of HIF-1α. There is also a pVHL-independent negative regulatory system that acts at the level of transactivation. In addition, the synthesis, degradation, and activity of HIF-1α are also regulated by an O2-independent system involving several cytokines and other signaling molecules [30]. Examples of this regulatory mechanism are the pathway including PI3K-AKT-mTOR, RAS/RAF/MEK/ERK kinase cascade, with phosphorylation of the CBP/p300 coactivator resulting in increased formation of the HIF-1α/p300 complex, the Hsp90 pathway and the Mdm2-p53 system, which is often altered in certain types of hypoxic tumors where low p53 levels are found, resulting in increased levels of HIF-1α [38,39].
HIF-1 acts as a transcription factor binding to 5′(A/G) CGTG- 3′ consensus sequences called hypoxia-responsive elements (HREs), which allows the activation of target genes [36] involved in the processes of tumor metastasis, angiogenesis, metabolic energy, cell differentiation and apoptosis [40,41]. Tumor cells often experience hypoxia due to a decrease in oxygen transport and diffusion and to an increase in O2 consumption. In fact, the intense proliferation of tumor cells causes an increased oxygen demand. Moreover, the distance between cells and the existing vascular system increases, making oxygen diffusion difficult and thus creating hypoxia. Hypoxia and a general imbalance in the distribution of oxygen within a solid tumor are typical features of neoplastic tissue and lead to more aggressive growth of the neoplasm. Oxygen deficiency leads to an increased expression of HIF-1α, which then activates angiogenesis, glucose metabolism, cell proliferation, invasion, and metastasis. Considering angiogenesis, the main HIF-1α target genes include VEGF, angiopoietin 1 and 2 and metalloproteases, leading to the formation of new, albeit unstructured, vessels within the tumor [42].
Beyond its roles in angiogenesis, HIF-1α also has several different roles in cancer progression. It triggers tumor cells invasion, acting in the epithelial–mesenchymal transition (EMT) process, increases cells proliferation and decreases apoptosis [42]. Moreover, HIF-1α is an important regulator of tumor cell metabolism, acting mainly on glucose catabolism, and favoring the development of an acidosic environment, stimulating fatty acid synthesis and glycogen synthesis (Warburg effect) [43].
The role of HIF-1α in HCC is certainly multifaceted as it is involved in several processes that influence carcinogenesis, such as vascularization, inflammation, infection by hepatotropic viruses, and changes in the microenvironment. The importance of HIF in these mechanisms has certainly stimulated research into the role that this marker might have in the treatment of liver cancer. Several in vivo studies highlight the importance of targeting the HIF pathway to inhibit tumor progression and to improve the efficacy of anti-VEGF therapies by controlling the hypoxic tumor microenvironment [44,45]. Indeed, long-term success of sorafenib treatment in HCC is limited due to the development of resistance caused by various mechanisms, including antiangiogenic effects and HIF-mediated cellular responses. Overexpression of HIF-1α and HIF-2α in HCC patients indicates a poor prognosis, prompting exploration of combined therapies targeting HIFs to overcome sorafenib resistance. Targeting both HIF-1α and HIF-2α shows potential as a more effective strategy than selective therapies as there is a strong correlation between the hypoxic microenvironment and sorafenib resistance [44]. Targeting HIF may limit the side effects caused by hypoxia induced by radiation or anti-angiogenic factor therapies, leading to clinically significant treatment improvements [46].
2.2. VEGF/VEGFR
The most well-known regulators of angiogenesis are the VEGF and VEGF receptors (VEGFRs) [47], which are fundamental for HCC development and progression. The ligands VEGF-A, VEGF-B, VEGF-D and VEGF-E belong to a family of structurally related dimeric proteins [48]. These growth factors (VEGF-A, VEGF-C, or VEGF-D) bind and stimulate VEGFR-2, which is expressed in nearly all endothelial cells [48]. VEGF-A is the most important isoform, responsible for angiogenesis and vascular remodeling. The ligand–receptor binding triggers downstream cellular pathways, involving many signal molecules (Y1213, Y1333, Sck, PLC-γ, VRAPAKT, FAK, p38 MAPK, eNOS, Src and PI3K), ultimately leading to formation of new tumor blood vessels within tumors which are essential for facilitating tumor development and progression [49,50]. VEGF expression, with its transcription regulated by the binding between HIF and hypoxia-responsive elements (HREs) [51], is modulated by tissue oxygen levels [52,53].
Increased levels of circulating VEGF have been observed in HCC [54] and have been demonstrated to be associated with accelerated disease progression and poorer prognosis [55,56]. In addition, VEGF seems to play a role in chemoresistance by acting on autophagy through NRP2 and mTOR [48]. These observations [55,56] provide support for the assessment of VEGF-pathway-directed therapies as a useful approach for treating HCC. Moreover, the groundbreaking survival results obtained with the combination atezolizumab + bevacizumab [23] confirm the importance of angiogenesis and support targeting the VEGF axis in HCC.
2.3. PDGF/PDGFR
The PDGF family consists of several ligands (PDGF-A, PDGF-B, PDGF-C, PDGF-D and PDGF-AB [57]) which bind to the tyrosine kinase PDGF receptor (PDGFR)-α and -β expressed on mesenchymal cells (fibroblast, smooth muscle cells, and pericytes). This interaction activates similar pathways to those stimulated by VEGF [57,58]. Binding of PDGF with their corresponding receptors leads to the activation of a signaling cascade which results in upregulation of VEGF and recruitment of perivascular cells. The relevance of PDGF/PDGFR pathways in human HCC is demonstrated by the fact that overexpression of PDGFR-α is associated with vessel density and worse prognosis [22]. Moreover, a shorter survival was demonstrated in HCC patients expressing PDGFR-α, PDGFR-β and VEGF [22]. Nevertheless, the inhibition of the PDGFR pathway as a target for anti-angiogenic therapy in HCC remains of uncertain clinical relevance. Although TKIs (sorafenib and others) also target PDGFR, these drugs also inhibit other pathways, so it is currently unclear what impact the inhibition of the PDGF pathway has on the overall clinical benefit.
2.4. FGF/FGFR
The FGF family includes several ligands that interact with four tyrosine kinase receptors (FGFR-1, -2, -3 and -4) [59]. FGFs and FGFRs are ubiquitously expressed, and among their various functions, they regulate cell growth and maintain VEGF-induced neovascularization [60]. During the initial phases of tumor growth, the cross-talk between FGF-2 and VEGF-A is able to induce neovascularization and boost tumor progression [61]. FGFs and VEGF-A are linked to enhanced capillarization of sinusoids [62], while FGF-induced integrin expression interacts with endothelial cells in the microenvironment, thereby modifying the essential cellular parameters required for angiogenesis. The resistance of advanced HCC to the VEGFR inhibitor sorafenib may be partially explained by this synergism between the FGF and VEGF pathways [63,64].
2.5. Angiopoietin/Tie Pathway
Angiopoietin 1 (Ang1) and 2 (Ang2) are ligands for the Tie2 receptor expressed on endothelial cells [65]. While vascular support cells widely express Ang1, Ang2 is only present at sites of vascular remodeling [66]. Indeed, Ang1 and Ang2 compete for their binding to Tie2, and these interactions modulate the pathway. Ang1 stabilizes the blood vessels, while Ang2 expression in areas of vascular remodeling competes with Ang1 for the interaction with Tie2, destabilizing blood vessel support cells. This is a necessary step to facilitate vessel proliferation induced by VEGF [66].
Patients with HCC showed high levels of Ang2, suggesting a central role in carcinogenesis, potentially together with VEGF [65]. Considering the importance of this pathway in the progression of HCC, some agents targeting Angiopoietins/Tie2 interaction alone or in combination with sorafenib have been tested in clinical practice [67], but any potential clinical benefit remains to be determined.
2.6. Endoglin (CD105)
Endoglin (CD105) expression is increased in actively dividing endothelial cells, including those found in liver cancers [68,69]. It functions as an accessory coreceptor of transforming growth factor-β (TGF-β), antagonizing its inhibitory effects, but it also modulates the transition of endothelial progenitor cells to mature epithelial cells [70].
Endoglin expression is associated with the HCC stage differentiation and aggressiveness, promoting invasion and metastatic spread by increasing VEGF expression [71]. Although intriguing, targeting this pathway for the treatment of HCC has an unclear clinical relevance.
This entry is adapted from the peer-reviewed paper 10.3390/medicina59061115
This entry is offline, you can click here to edit this entry!