The suppression of excessive immune responses is necessary to prevent injury to the body, but it also allows cancer cells to escape immune responses and proliferate. Programmed cell death 1 (PD-1) is a co-inhibitory molecule that is present on T cells and is the receptor for programmed cell death ligand 1 (PD-L1). The binding of PD-1 to PD-L1 leads to the inhibition of the T cell receptor signaling cascade. PD-L1 has been found to be expressed in many types of cancers, such as lung, ovarian, and breast cancer, as well as glioblastoma. Furthermore, PD-L1 mRNA is widely expressed in normal peripheral tissues including the heart, skeletal muscle, placenta, lungs, thymus, spleen, kidney, and liver. The expression of PD-L1 is upregulated by proinflammatory cytokines and growth factors via a number of transcription factors. In addition, various nuclear receptors, such as androgen receptor, estrogen receptor, peroxisome-proliferator-activated receptor γ, and retinoic-acid-related orphan receptor γ, also regulate the expression of PD-L1.
1. Introduction
Immunity is a system that recognizes foreign substances, such as pathogenic microorganisms and cancer cells, as “non-self” and eliminates them from the body. For the immune system to function properly, the activation of the immune response is necessary to eliminate “non-self” cells, in addition to the suppression of the immune response to prevent damage to “self” cells caused by an excessive immune response. T cells play an important role in the specific immune response against cancer cells. T cell activation requires antigen presentation by antigen-presenting cells and co-signaling through the interaction of molecules expressed on the antigen-presenting cells and the T cells. Among these molecules expressed on the T cell surface, those that activate the T cells are called co-stimulatory molecules, which transmit activation signals, while those that inhibit the T cells are called co-repressive molecules, which transmit inactivation signals. T cells receive the antigen presentation information from the antigen-presenting cells via major histocompatibility complexes (MHCs), and T cells make contact with the MHCs molecules on the antigen-presenting cells via the T cell receptor (TCR) and the cluster of differentiation 4/8 (CD4/8), which facilitates antigen recognition [
1]. When co-stimulatory molecules on T cells bind to ligands on the antigen-presenting cells, the T cells are activated, and when the co-inhibitory molecules on T cells bind to the ligands on the antigen-presenting cells, T cell activity is suppressed. In this immune cell regulatory mechanism, the function of suppressing an excessive immune response is necessary to prevent injury to the body, but it also allows cancer cells to escape from the immune response and proliferate [
2]. Programmed cell death 1 (PD-1) is a co-inhibitory molecule on T cells and an immune checkpoint molecule. The receptors for PD-1 are programmed cell death ligand 1 (PD-L1) and PD-L2 [
3,
4,
5]. The binding of PD-1 and PD-L1 leads to the inhibition of the TCR signaling cascade. PD-L1 mRNA is widely expressed in normal peripheral tissues, including the heart, skeletal muscle, placenta, lungs, thymus, spleen, kidney, and liver. However, PD-L1 mRNA is not detectable in the brain, colon, and small intestine by Northern blot hybridization [
5,
6]. In addition, PD-L1 has also been found to be expressed in many types of cancers, including melanoma, glioblastoma, and renal, lung, ovarian, and breast cancer [
7]. Thus, the inhibition of PD-1 and/or PD-L1 by antibodies is an immunotherapy strategy for cancer treatment [
8]. The expression of PD-L1 is upregulated by proinflammatory cytokines, such as interferon-γ, tumor necrosis factor-α, and interleukin-6 (IL-6), and growth factors, such as epidermal growth factor and hepatocyte growth factor, as well as gases, such as oxygen (hypoxia) and nitric oxide, via several transcription factors [
9,
10,
11]. However, various nuclear receptors also regulate the expression of PD-L1.
2. Regulation of PD-L1 Expression by Nuclear Receptors
Various transcription factors, such as activator protein 1 (AP-1), NF-κB, STAT1, STAT3, IRF1, IRF3, and hypoxia-inducible factor 1, are activated by a variety of stimuli, such as growth factors, cytokines, and hypoxia, to regulate the gene expression of
PD-L1 [
49]. However,
PD-L1 gene expression is not only controlled by transcription factors but also various nuclear receptors (
Figure 2).
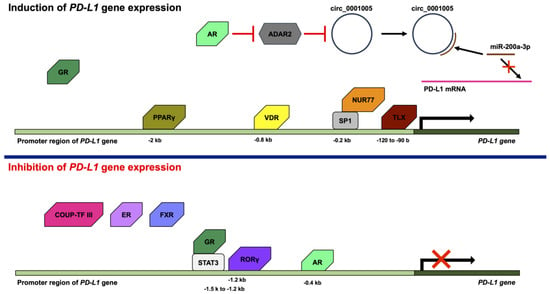
Figure 2. Effects of nuclear receptors on PD-L1 expression. Nuclear receptors positively or negatively regulate the PD-L1 gene expression. The androgen receptor (AR), glucocorticoid receptor (GR), NUR77, peroxisome-proliferator-activated receptor γ (PPARγ), TLX (homolog of the Drosophila tailless gene), and vitamin D receptor (VDR) induce the PD-L1 gene expression. AR decreases expression of adenosine deaminases, which act on RNA 2 (ADAR2), leading to the increases in the expression of circ_0001005. Circ_0001005 increases the expression of PD-L1 by competitive binding to microRNA-200a-3p (miR-200a-3p). GR is associated with the induction of PD-L1 gene expression. NUR77 binds to the −0.2 kb promoter region of the human PD-L1 gene with SP1. PPARγ binds to the −2 kb promoter region of the human PD-L1 gene. TLX binds to the −120 to −90 b promoter region of the human PD-L1 gene. VDR binds to the -0.8 kb promoter region of the human PD-L1 gene. COUP-TF III, estrogen receptor (ER), farnesoid X receptor (FXR), and retinoic-acid-related orphan receptor γ (RORγ) inhibit the PD-L1 gene expression. Furthermore, AR and GR also inhibit the PD-L1 gene expression. AR inhibits the PD-L1 gene expression by binding to the −0.4 kb promoter region of the human PD-L1 gene. COUP-TF III, ER, and FXR are associated with the inhibition of PD-L1 gene expression. GR binds to the −1.5 k to −1.2 kb promoter region of the human PD-L1 gene with signal transducer and activator of transcription 3 (STAT3). RORγ binds to the −1.2 kb promoter region of the human PD-L1 gene.
AR is a member of the steroid hormone nuclear receptor group that consists of ER, GR, the mineralocorticoid receptor, and the progesterone receptor [
55,
56,
57]. Although AR was found to be a ligand-activated nuclear receptor, AR also functions in a ligand-independent manner [
58,
59,
60]. The overexpression of AR results in the decreased expression of PD-L1, and the knockdown of AR by small hairpin RNA (shRNA) leads to the increased expression of PD-L1 in human hepatocellular carcinoma cells. AR binds androgen response element (ARE) in the −0.4 kb promoter region of the human PD-L1 gene. In addition, the anti-PD-L1 antibody treatment of mice implanted with Hep1-6-PCDH (AR-) or Hep1-6-AR (AR+) tumor cells resulted in smaller tumors in mice implanted with Hep1-6-PCDH (AR-) cells than in mice implanted with Hep1-6-AR (AR+) cells [
61]. In addition, the treatment of 84E7 cells (AR-expressing thyroid cancer cells) with dihydrotestosterone (DHT), which is an agonist of AR, resulted in the suppression of PD-L1 expression. In contrast, the treatment of 8505C cells (thyroid cancer cells that do not express AR) with DHT did not alter the expression of PD-L1. Treatment of 84E7 cells with Flutamide, a selective AR antagonist, recovered from the DHT-suppressed PD-L1 expression. In addition, AR binds to the PD-L1 gene and downstream of the PD-L1 open reading frame [
62]. By contrast, the overexpression of AR resulted in an increase in the expression of PD-L1, and the knockdown of AR by shRNA led to a decrease in the expression of PD-L1 in bladder cancer cells. The increase in the expression of PD-L1 by AR is dependent on adenosine deaminases, which act on RNA 2 (ADAR2), and circ_0001005, which is a circular RNA (circRNA). AR decreases the expression of ADAR2, and the knockdown of ADAR2 by shRNA leads to the increases in the expression of circ_0001005. Circ_0001005 increases the expression of PD-L1 by competitive binding to microRNA-200a-3p (miR-200a-3p) [
63]. The circRNA is a single-stranded RNA that forms a covalently linked loop between its 5′ and 3′ ends. circRNA is possibly generated by lariat-driven circularization, intron pairing-driven circularization, or RNA-binding protein-driven circularization. circRNAs absorb microRNAs (miRNAs) to act as sponges for miRNAs and inhibit the transcription of the miRNA target gene [
64]. miRNAs are short non-coding RNAs, which are not translated into proteins. miRNAs basically consist of approximately twenty bases and bind to mRNAs, leading to the degradation of mRNAs or the inhibition of translation from mRNA. Thus, miRNAs play a role in the intrinsic RNA silencing [
65]. The 3′-untranslated region of PD-L1 contains the target sequence of miR-200 [
66].
COUP-TF III, which is also known as nuclear receptor subfamily 2 group F member 6 (NR2F6) or v-erbA-related protein, is an orphan nuclear receptor [
67,
68]. Nr2f6 knockout mice exhibit a substantially higher expression of PD-1 and PD-L1. In addition, Nr2f6 knockout mice are resistant to the generation of sarcoma induced by methylcholanthrene and sarcoma transplantation [
69]. Moreover, anti-PD-L1 antibody treatment reduces the tumor growth of Nr2f6 knockout mice with sarcoma transplantation [
69,
70]. In addition, the PD-L1 expression levels are increased in the CD4
+ and CD8
+ T cells from Nr2f6 knockout mice [
69].
ER is also one of members of the steroid hormone nuclear receptor group [
71]. Although ER is activated by ligands, such as estrogen, ER also functions in a ligand-independent manner [
60,
72,
73]. The PD-L1 mRNA expression levels in ERα-positive breast cancer cells are lower compared to the PD-L1 mRNA expression levels in ERα-negative tumors from patients with breast cancer [
74]. In addition, MCF7 cells, which are ER-positive but human epidermal growth factor receptor 2 (HER2)-negative cells, also express low levels of PD-L1 mRNA. Thus, ER negativity is proportional to high PD-L1 mRNA expression. Moreover, treatment with estradiol repressed the expression of PD-L1 mRNA [
75]. In addition, the knockdown of ERα by small interfering RNA (siRNA) results in the upregulation of PD-L1 in MCF7 cells. However, knockdown of ERβ by siRNA did not upregulate the expression of PD-L1 in MCF7 cells [
76]. The expression of ER is associated with the expression of IL-17E, which is also known as IL-25. IL-17E leads to a reduction in IL-17A, C, and F [
77]. IL-17A enhances the expression of PD-L1 in triple-negative (ER-negative, PR-negative, and HER2-negative) breast cancer cells (MDA-MB-231 and SKBR-3 cells) via the phosphorylation of extracellular signal-regulated kinases [
78]. Comparing the breast cancer cell lines, it is noted that the ER-positive breast cancer cell lines (MCF7 and T47D) do not express PD-L1, but the ER-negative breast cancer cell lines (BT-20, BT-549, HCC38, MDA-MB-231, and SKBR-3) do express PD-L1. However, AU565 and HCC1143, which are ER-negative breast cancer cell lines, do not express PD-L1 [
75,
78,
79]. In addition, the treatment of MC38-colon-tumor-injected mice with estrogen and anti-PD-L1 antibody significantly reduced MC38 tumor growth compared to estrogen or anti-PD-L1 antibody alone [
80].
Farnesoid X receptor (FXR) forms a heterodimer with RXR and the ligands for FXR are bile acids. FXR regulates various genes related to the metabolic, immune, and nervous systems [
81]. The expression of FXR and PD-L1 in non-small cell lung cancer (NSCLC) and hepatocellular carcinoma cells is inversely correlated [
82,
83,
84]. The knockdown of FXR by siRNA leads to the promotion of PD-L1 expression in NSCLC cells. In addition, FXR binds to the promoter region of the PD-L1 gene [
83].
GR positively or negatively regulates the expression of a wide variety of genes [
85,
86]. The knockdown of GR by shRNA results in a reduction in PD-L1 in SU86.86 cells, which are pancreatic cancer cells. Moreover, the treatment of SU86.86 cells with dexamethasone induces the expression of PD-L1, and several glucocorticoid response elements have been found in the promoter region of PD-L1. In addition, the expression of GR is related to the expression of PD-L1 and a low survival rate in patients with pancreatic cancer [
87]. However, treatment with dexamethasone leads to a decrease in the expression of PD-L1 in other cancer cell lines, including SGC-7901 (human gastric cancer cell line), MKN-45 (human gastric cancer cell line), SMMC-7721 (human hepatocarcinoma cell line), and BxPC3 (human pancreatic cancer cell line). Dexamethasone enhances the formation of GR and the STAT3 complex. The human PD-L1 gene is located at chr9:5450503-5470566. The GR/STAT3 complex binds to the binding sites of STAT3 in the promoter region of human PD-L1 (ch9:5449027-5449342), which is from −1.5 k to −1.2 kb in the promoter region of the human PD-L1 gene, and inhibits PD-L1 gene expression by STAT3 [
88].
NUR77, which is also known as nuclear receptor subfamily 4 group A member 1 (NR4A1), is an orphan nuclear receptor. NUR77 plays an important role in the regulation of cell proliferation, apoptosis, and the metastasis of various tumors, including breast, colorectal, and liver cancers [
89,
90]. It has been reported that 1,1-Bis(3′-indolyl)-1-(p-hydroxyphenyl) methane (CDIM-8, which is also known as DIM-C-pPhOH) and 1,1-bis(3′-indolyl)-1-(3-chloro-4-hydroxy-5-methoxyphenyl)methane (Cl-OCH
3) act as NR4A1 antagonists [
91]. Treatment with CDIM-8 or Cl-OCH
3 leads to a decrease in the expression of PD-L1 in breast cancer cell lines (Hs578T, SUM159PT, MDA-MB-231, and 4T1), a lung cancer cell line (A549), a colon cancer cell line (SW480), and a kidney cancer cell line (786-0). Moreover, the knockdown of NR4A1 by siRNA leads to the decreased expression of PD-L1 in MDA-MB-231 and 4T1 cells [
92,
93]. NR4A1 binds to the −0.2 kb promoter region of the human PD-L1 gene with SP1, and CDIM-8 and Cl-OCH
3 inhibit the binding of NR4A1 to the promoter region of the human PD-L1 gene [
92]. In addition, NR4A1 binds to the AP-1 binding site and prevents AP-1-induced gene expression by blocking transcription by AP-1 [
94].
PPARγ forms a heterodimer with RXR and binds to the PPAR response elements (PPAREs). PPARγ regulates various genes associated with metabolism related to lipid, glucose, and cholesterol [
95,
96]. Treatment with PPARγ ligands, such as rosiglitazone and pioglitazone, induces the expression of PD-L1 in HT29 and HCT116 cells, which are gastrointestinal adenocarcinoma cell lines. Furthermore, PPARγ with the treatment of rosiglitazone binds to PPAREs in the proximal −2 kb promoter of the human PD-L1 gene [
97]. Furthermore, PPARγ binds to PPREs in the PD-L1 promoter and treatment with rosiglitazone induces the expression of PD-L1 in tumor organoids from microsatellite-stable positive patients with colorectal cancer. Moreover, treatment with rosiglitazone and IFN-γ reduces the cell viabilities of tumor organoids in the presence of the wild-type anti-PD-L1 antibody. It is speculated that the wild-type anti-PD-L1 antibody blocks the PD-L1 induced by rosiglitazone and IFN-γ, but also bridges the Fc receptor on NK/T-like cells and PD-L1 on tumor organoids, promoting the attack of tumor organoids by NK/T-like cells [
97]. In addition, treatment with GW9662, which is a PPARγ antagonist, suppresses the expression of PD-L1 in 3T3-L1-cell-induced adipogenesis [
98].
RORγ, which is also known as retinoic-acid-related orphan receptor C (RORC), is an orphan nuclear receptor and plays an important role in the immune system [
99,
100,
101]. The overexpression of RORγ in human 5637 and UC3 cells, which are bladder cancer cell lines, leads to a reduction in PD-L1 expression and the suppression of the PD-L1 promoter activity. RORγ binds to the ROR response element in the proximal −1.2 kb promoter of the human PD-L1 gene. In addition, the overexpression of RORγ represses the expression of the STAT family proteins, including STAT1 and STAT3. Moreover, the overexpression of RORγ leads to the phosphorylation of STAT3 and suppresses the binding activity of STAT3 [
102].
Homologue of the drosophila tailless gene (TLX), which is also known as nuclear receptor subfamily 2 group E member 1 (NR2E1), is an orphan nuclear receptor [
103]. However, a recent study demonstrated that oleic acid may be the ligand for TLX [
104]. The expression of TLX is correlated with the expression of PD-L1 in glioma tissue in patients with glioma. In addition, the knockdown of TLX by shRNA leads to the repression of PD-L1 in the human glioblastoma cell line A1235. TLX binds to the proximal −122 to −90 promoter of the human PD-L1 gene [
105].
VDR forms a heterodimer with RXR and binds to the VDR response elements (VDREs). VDR plays a crucial role primarily in the regulation of calcium homeostasis [
106]. PD-L1 is upregulated by the 1,25-dihydroxyvitamin D treatment of human squamous cell carcinoma cells (SCC25 and SCC4) and human myeloid cells (THP-1 cells). Furthermore, VDR binds to the VDRE in the proximal −0.8 kb promoter region of the human PD-L1 gene in SCC25 cells and THP-1 cells that were differentiated into macrophages. In addition, the treatment of a co-culture of SCC25 cells and T cells from a healthy donor with 1,25-dihydroxyvitamin D reduces T cells from the healthy donor, and anti-PD-L1 antibody partially or completely prevents the reduction in T cells [
107].
This entry is adapted from the peer-reviewed paper 10.3390/ijms24129891