Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Oncology
Breast cancer cells travel via the bloodstream to the bone before the cancer is detectable in the breast. These disseminated cells are resistant to adjuvant chemotherapy and hormone therapy administered for the very purpose of eliminating them. They recur steadily for more than 20 years, resulting in incurable diseases. The bone marrow location, or niche, which normally provides a nest for blood-forming cells to enable them to generate blood for the entire lifetime of an individual, also protects these disseminated tumor cells and places them into a state of quiescence called dormancy.
- dormancy
- micrometastases
- hematopoietic niche
- bone marrow microenvironment
1. Introduction
More than 43,000 women in the US die from breast cancer (BC) every year, primarily from metastatic disease [1]. However, BC cells metastasize to the bone marrow (BM) before primary tumors can be detected and are found in the BM of 27–40% of newly diagnosed patients with localized primary disease [2,3]. Once they arrive at the BM, most micrometastases are killed by the hostile microenvironment; nevertheless, some of them enter a state of dormancy [4]. Dormant cells have cancer stem cell characteristics [5] and are resistant to adjuvant chemotherapy administered for the distinct purpose of eliminating them [6,7]. The metastatic niche contributes significantly to this resistance [8,9].
2. Breast Cancer Metastasis and Dormancy in the Bone Marrow
2.1. Hematogenous Transit of Cancer Cells to the BM HSC Niches
The homing of cancer cells to the BM is reported to approximate that of HSCs [24]. It is mediated through the C-X-C motif chemokine receptor 4 (CXCR4), the receptor for stromal cell-derived factor-1/C-X-C motif chemokine 12 (SDF-1/CXC ligand (CXCL)12) [25,26,27] and through annexin II [28], which is required for hematopoietic stem cell transplants [29]. Annexin II serves as an anchor for CXCL12 to localize HSCs [30] and cancer cells [31] to the niche. Homing also involves cadherin-11 [32,33], osteopontin [34], connective tissue growth factor (CTGF) [34], and Runt-related transcription factor 2 (RUNX2) [35]. Cadherin-11 induces the expression of the gamma-carboxyglutamic acid (Gla) domain-containing protein 6 (GAS6) receptors AXL tyrosine kinase (AXL), skywalker (Sky) and Mer proto-oncogene tyrosine kinase (Mer), which induce dormancy in hematopoietic stem cells [36].
Disseminated tumor cells (DTCs) in the BM interact with a wide array of cell types, proteins, proteoglycans, growth factors and cytokines endemic to the hematopoietic microenvironment, which, together with its biophysical and bioenergetic characteristics, regulate dormancy and participate in reawakening [37]. The BM hematopoietic microenvironment is made up of a complex network of cells consisting of mesenchymal cells of different lineages and degrees of stemness, osteogenic cells, chondrocytes, adipocytes, neuroglial cells, hematopoietic lineage cells, including megakaryocytes and macrophages, cells of the sympathetic nervous system and a network of endothelial cells that include cells lining the sinusoids, arterioles and transition zones [38]. These data indicate the existence of two primary niches that maintain HSC dormancy, the preosteoblast endosteal-lining niche [39] and a more centrally located parasinusoidal endothelial niche, with some endothelial cells also residing near the endosteum [40]. The two positions have been reconciled by arguments that the endosteal niche is also vascularized [41], that the niches are in close proximity and that, because of the newly recognized heterogeneity in the HSC population gleaned from single-cell sequencing, different niches may provide support or imprint distinct HSC states for differently primed HSCs [38,42,43,44,45].
The endosteal niches consist of osteoblasts embedded in the bone matrix and preosteoblasts that are in contact with HSCs and Nestin-GFP+hi mesenchymal stem cells (MSCs), which regulate HSC maintenance. Macrophages also inhabit the endosteum and help maintain the HSC niche. The sympathetic nervous system maintains HSCs through nerve fibers in the endosteum. Structural elements of the endosteum also support the dormancy of HSCs, including fibronectin and heparan sulfate proteoglycans (HSPGs) that serve as reservoirs for fibroblast growth factor (FGF)-2, which is important in HSC dormancy (Figure 1). I have discussed these members of the HSC niche below.
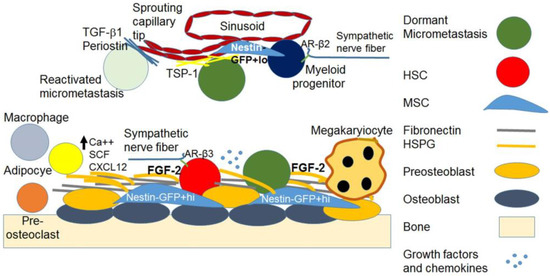
Figure 1. The bone marrow hematopoietic and dormant metastatic breast cancer niches. The image depicts simplified representations of the endosteal and parasinusoidal niche containing the HSC, the dormant micrometastasis, and the supporting cellular, structural and soluble elements. Growth factors and chemokines listed in the text are depicted as a cluster of light blue dots.
The true, rare, dormant and undifferentiated HSCs reside on preosteoblasts in the endosteal stromal and preosteoblast stem cell niches, which support their quiescence and self-renewal [46,47]. Once they acquire a lineage marker, they move to the sinusoidal endothelium where they can be tapped to differentiate into myeloid lineages [46,47].
Only immature osteoblasts support the dormancy of HSCs [48], whereas ossified, alkaline phosphatase (ALP)-expressing differentiated osteoblasts no longer support HSCs [49,50,51,52]. Signaling through parathyroid hormone (PTH) and interleukin (IL)-6, and adhesion to vascular cell adhesion molecule (VCAM)-1 and activated leukocyte cell adhesion molecule (CD166) in osteoblast precursors is necessary for appropriate HSC maintenance and lymphoid differentiation [49,50,51]. An array of adhesion molecules, growth factors and chemokines interact to maintain HSCs in a quiescent state in their niche and to mobilize them as needed for recruitment to the vascular niche [47]. These include Angiopoietin-1, Tie-2 and N-cadherin, which are associated with quiescence and cell-cycle control and adhesion molecules, including very late antigen (VLA)-4, lymphocyte function-associated (LFA)-1, osteopontin and integrins [47]. These molecules, along with CXCL12 (SDF-1) on stromal cells, its receptor CXCR-4 on HSCs, matrix metalloproteinase (MMP)-9, MMP-2 and stem cell factor (Kit Ligand) induced by granulocyte-colony stimulating factor (G-CSF), SDF-1, FGF-4, vascular endothelial growth factor (VEGF) and placental growth factor (PLGF), are required for HSC recruitment and mobilization [47]. Calcium in the endosteal niche is also important for the support of HSCs [53]. HSC maintenance and quiescence, hematopoietic reconstitution and protection from aging-associated DNA damage also depend on interactions with periostin via integrin αv and inhibition of focal adhesion kinase (FAK)/phosphoinositide 3-kinase (PI3K)/Ak strain transforming (AKT) signaling, leading to an increase in p27Kip1 [54].
The sympathetic nervous system β2 adrenergic signaling releases HSCs from the osteoblastic niche by inducing the transcription of the calcium-regulating hormone vitamin D receptor (VDR) and its downstream gene receptor activator of nuclear factor kappa-Β ligand (RANKL), an effect stabilized by 1,25 dihydroxy-vitamin D3 [55]. While the mean extracellular calcium ion concentration [Ca++]e in the BM is 1.0 ± 0.54 mM, which is not significantly different from that in the blood serum, the [Ca++] in the location of the HSCs is 1.5 ± 0.57 mM and significantly increases with aging to support myeloid clonal expansion [56].
Aging and senescence of HSC niches result in changes in the character, makeup, location and differentiation dynamics of HSCs by regulating niche remodeling [42]. Aging induces a functional decrease in adrenergic receptor (AR)-β3 activation and increases AR-β2 (ADRB2) activation [42,47,57]. This induces HSC migration from osteoblasts to the sinusoids, skewing hematopoiesis towards myeloid differentiation, megakaryocyte and platelet production, and decreases endosteal cells, resulting in lymphoid deficiency [42,47,57]. Aging stroma results in a decrease in endosteal and in an increase in sinusoidal Nestin-GFP+hi cells, events that also play a role in the myeloid skewing of hematopoiesis. The movement of Nestin-GFP+hi cells, which give rise to all mesenchymal lineages, including osteoblasts and adipocytes, to the sinusoidal space induces the migration of Jagged-stained cells and associated HSCs to the sinusoids [42,58].
There is a global decrease in osteoblastogenesis and an increase in adipocytogenesis with aging, ovariectomy and other causes of osteoporosis or disease [59,60,61,62]. Indeed, reinforcing the role of adipocytes in the loss of dormancy, perivascular cells express an adipocyte-skewed expression profile that promotes proliferation [63]. Mature adipocytes activate extracellular receptor kinase (ERK or MAP kinase) signaling in multiple myeloma cells [64]. Overall vascular density and leakiness increase and sinusoidal notch receptor (Notch) activity decreases with aging [42]. These effects are accompanied by an overall increase in inflammation and secretory senescence, characterized by increased levels of inflammatory cytokines IL-1, IL-3, IL-6, tumor necrosis factor (TNF)α, interferon (INF)γ and transforming growth factor (TGF)-β, which regulate myeloid skewing [42].
The role of osteoblasts in the maintenance of the undifferentiated quiescent HSC state is supported by experiments in which a preosteoblast knockout was sufficient to induce leukemia [65,66,67], and its replacement restored normal hematopoiesis [68]. Many adhesion molecules, growth factors and chemokines interact to maintain HSCs quiescence in their niche and mobilize them to the vascular niche as needed [47,49,50,51]. These include bone morphogenic proteins (BMPs), TGF-β2 [46] and FGF-2 [69,70,71]. FGF-2 is important for the maintenance [69], self-renewal [70,71,72] and myeloid differentiation of HSCs [73,74], but blocks erythroid and B lymphocyte differentiation [75] and myeloid differentiation at high concentrations [76].
2.2. The Metastatic BM Niches and DTC Dormancy Signaling
Most metastases die in the hostile microenvironment of the BM, but some survive and enter a state of dormancy [77]. Early arriving DTCs that survive the microenvironmental effects generate a metastatic niche, combine with late metastases and potentially remain quiescent or in an ultra-slow cycling mesenchymal state in the HSC niche [78,79,80] for periods lasting up to decades [46,81].
The fate of the cancer cells depends on the opposing efforts of the microenvironment and the cancer cells. The microenvironment endeavors to suppress the cancer cells, while the cancer cells exert their efforts to turn MSCs and fibroblasts into cancer-promoting cells [82]. The cancer cells attempt to generate a pre-metastatic niche with the potential to support cancer cell colonization through the modulation of MSC through microvesicles [83] and through the secretion of inflammatory cytokines, which recruit BM-derived cells and form an inflammatory milieu that supports colonization [84]. They also secrete factors that enhance bone resorption, such as lysyl oxidase (LOX), a collagen crosslinking enzyme produced primarily by hypoxic ER− cancer cells, parathyroid hormone-related peptide (PTHrP), osteopontin (OPN) and CC-chemokine ligand (CCL)-2, directly promoting bone resorption and extracellular matrix (ECM) remodeling, making the niche more permissive to DTCs [85]. However, metastatic cancer cells also process structural proteins such as fibronectin in the microenvironment, which in turn induce quiescence and survival signaling in the cancer cells [81]. Ultimately, the scarcity of micrometastases is the most likely reason why the cancer cells’ attempts at modifying the niche to promote cancer growth are overwhelmed by the collective suppressive effects of the cellular, structural and soluble factors of the niche [82]. DTCs in the BM interact with a wide array of cell types, proteins, proteoglycans, growth factors and cytokines endemic to the hematopoietic microenvironment, which, together with their biophysical and bioenergetic characteristics, regulate dormancy and eventually participate in reawakening [37].
Cancer cell dormancy in a niche can be considered an adaptive state, guided by the thermodynamics of local energy minima, mechanical confinement [86] and hypoxia [87]. The preparation of the metastatic niche may be aided by the presence of VEGF receptor (VEGFR)1+ hematopoietic progenitor cells [88], as well as the deposition of extracellular matrix by micrometastases [89]. In the metastatic HSC niche, BC cells interact with cellular, structural and soluble factors to initiate dormancy [88,90,91,92,93,94], including NG2+/Nestin+ mesenchymal stem cell-initiated TGF-β2 and BMP7 signaling [95]. Cells include MSCs, fibroblasts, osteoblasts, adipocytes, Nestin+ endothelial cells, T-cells and macrophages. Structural factors include fibronectin, p-selectin, thrombospondin and HSPG. Soluble factors include Bmp4, Bmp6, Bmp7, kit ligand, TGF-β1 and β2; Dickkopf-related protein 1 (Dkk1) and Dkk3, thombospondin2 (Thbs2) found in the BM secretome [96,97] and FGF-2 [88,90,91,92,93,98]. FGF-2 is deposited on stromal HSPGs [99], which are needed for FGFR dimerization [100] and are able to induce dormancy [101]. Fibronectin, an integral element of the endosteum [102], also induces dormancy [103,104] and can prevent transformation [105]. Signaling initiated by structural proteins in the BM also depends on their variable structural organization [103], tensile strength and mechanical signaling [106].
Dormant BM micrometastases have marked genetic heterogeneity [107,108]. However, most express the hyaluronan receptor CD44 and about half of the cells are also CD24−, identifying them as having tumor-initiating characteristics that enable them to regrow into breast tumors [109] and express a stem cell program [110]. Signaling initiated by osteoblast interactions seems to maintain tumor-initiating properties in DTCs [111].
Maintenance of stemness was also reported to be mediated by FGFR2 [112] and FGFR-initiated signaling through Akt/Sry-related HMG-box (Sox)2 [113], stem cell-like chromatin rearrangement through the inhibition of cyclin-dependent kinase (CDK)4/6 and upregulation of programmed cell death protein (PD)-1 [114,115], protecting dormant cells from immune elimination. A key niche factor for inducing and maintaining ER+ BC dormancy is FGF-2, which is synthesized and exported by stromal fibroblasts and subsequently deposited on HSPGs overlying the stroma [98,99]. FGF-2 also plays a key role in the maintenance of HSC dormancy, as noted above [112,113]. HSPGs are necessary for the dimerization of FGF receptors [100] and for maintenance [116], multipotency [117] and osteogenic differentiation of MSCs [118]. Similarly to its role in HSCs, FGF-2 also supports the dormancy of hormone receptor-positive BC cells through dual signaling by FGF-2-induced re-expression of integrin α5β1, which binds to microenvironmental fibronectin [98,119,120,121]. FGF-2 inhibits breast cancer cell proliferation and response to chemotherapy through the activation of ERK [122], phosphoinositol-3 kinase (PI3K) [98,119] and intracellular TGF-β-mediated upregulation of cyclin-dependent kinase inhibitors p21Waf1, p27Kip1 and 15INK4b [123,124], inactivation of CDK2 and CDK4, and dephosphorylation of retinoblastoma protein (Rb) [123], mechanisms which have been confirmed in subsequent investigations [114,115]. Dormant cells maintain a characteristic, large, spread out, non-motile epithelial phenotype through dual FGF-2 and fibronectin-activated integrin α5β1 signaling [120]. The phenotype is due to the inhibition of Ras homology family member (Rho)A by the Rho GTPase activating protein (Gap) 26 (GRAF), a resulting cortical actin rearrangement and an omnidirectional activation of FAK [120]. Fibronectin, which is deposited abundantly in the BM microenvironment [102], suppresses the malignant phenotype [103,105] and collaborates with integrin α5β1 to establish the premetastatic niche [81]. ER− cells are not inhibited by FGF-2 and do not utilize the fibronectin-FGF-2 dual signaling model to become dormant [98]. However, stromal MSCs do inhibit ER− BC cells in a transwell co-culture model through the transfer of micro (mi)RNAs 127, -197, -222 and -223 [125,126,127] or SDF-1a [128] and decrease in CXCL12 levels [126]. This interaction is reciprocal, as metastatic breast cancer cells in the bone marrow microenvironment participate in remodeling the niche to sustain their dormancy [129]. In addition to its role in BC [130], FGF-2 also promotes stemness in benign prostate cells [112] and pancreatic cancer [113], and induces dormancy in ER+ BC cells [98]. Quiescent cancer micrometastases express dormancy signatures [78,79,131] similar to those modulating normal stem cell quiescence [78,80]. FGF-2 acts in concert with structural proteins in the microenvironment, where dormant micrometastases become anchored in place by binding to microenvironmental proteins and cellular components [98,104].
The MSC niche, the vascular niche and the immune niche provide support for metastatic BC cell survival and dormancy through a variety of mechanisms (Table 1) [46]. Metastatic cells survive in the BM hematopoietic microenvironment in close proximity to stromal cells in the endosteum, where they occupy the hematopoietic stem cell niche [132], as well as in the perivascular endothelium [8]. The mesenchymal stem cell niche activates multiple signaling pathways in dormant cells via receptors Mer tyrosine kinase (MERTK), AXL and its ligand GAS6 [36,133], TGF-β2 through TGF-β receptor 3 and BMP receptor 2 via SMAD family members (SMAD)1 and 5, basic helix–loop–helix family member E41 (DEC2), the metastasis suppressor gene N-Myc downstream-regulated (NDRG)1, BMP4 and 7 through BMP receptor (R)2, activated p38 MAP kinase (p38) [90] and its downstream target mitogen- and stress-activated kinase 1 (MSK1) [134], activin receptor-like kinase (Alk)5 [135], inactivated ERK, and cyclin-dependent kinase inhibitors p21Waf1 and p27Kip1 [46,94,136]. Other stem cell niche signals also regulate dormancy [88,90,91,93], potentially by antagonizing oncogene signaling [92].
The BM is a hypoxic environment [137], a factor implicated in the induction of dormancy [138] by repressing the leukemia inhibitory factor (LIF)-signal transducer and activator of transcription (STAT)3. Primary tumor hypoxia presets primary tumor cells with a program supporting dormancy, which manifests after dissemination to the metastatic niche [139]. Redox signaling in the microenvironment also generates enabling conditions for dormancy signaling, remodeling of the microenvironment, reprogramming of DTC dormancy signaling and maintenance of epithelial-mesenchymal transition (EMT) and stemness [140]. Microenvironmental redox signaling also generates therapeutic resistance in dormant cells through vigorous induction of antioxidant mechanisms to counter cytotoxin-induced oxidative stress, apoptosis, autophagy and oncogenic bypass signaling [140]. Conversely, redox signaling can also play a role in reawakening [140].
Other factors involved in inducing dormancy are retinoic acid, leukemia inhibitory factor (LIF), wingless-related integration site (Wnt) family members, miR-126 and DNA methylation (reviewed by Risson et al., 2020) [94]. Stroma also produces exosomes overexpressing miR-23b [125] or other miRNAs [126,127] that are transferred to DTCs, which also endow dormancy signaling [96,125,126,141]. Wnt5a non-canonical Wnt signaling induces dormancy in prostate cancer cells in vitro and in vivo in the BM osteoblast niche in a reversible manner via receptor tyrosine kinase-like orphan receptor 2 (ROR2)-activation of siah E3 ubiquitin protein ligase 2 (SIAH2) expression, which represses canonical Wnt/β-catenin tumor stem cell and tumor progression signaling [142]. Wnt family members regulate MSCs in their niche in the BM stroma, where Wnt5a localizes with cells that are leucocyte common antigen (CD45)+, which are transmembrane protein tyrosine phosphatases located on most hematopoietic cells, and CD45− mesenchymal stem cell marker (STRO-1)+ mesenchymal progenitor cells, whereas canonical Wnt is associated with the underlying stroma matrix [143]. Wnt3a expands the pool of MSCs capable of generating colony-forming unit-fibroblasts (CFU-F) and CFU-osteoblasts (O), whereas Wnt5a maintains the pool of cell numbers, CFU-Fs and CFU-Os, suggesting a potential dual role of Wnt5a in the maintenance of MSCs in the BM and in enhancing osteogenesis [143].
The BM microenvironment has a low oxygen tension, which predisposes cancer cells to fuse with MSCs and other cells in the microenvironment, and, in fact, the fusion of BC cells with MSCs can induce dormancy [144], as well as a spectrum of other functions in BC cells [145]. One study suggests that cancer cells cannibalize stromal mesenchymal cells to become dormant [146]. The BM interstitial pH ranges from 6.7 to 7.5 (7.0–7.3 within a 10% to 90% confidence interval), with a mean value of 7.1, which is slightly more acidic than the blood serum that is close to 7.4 [56]. The BM oxygen tension is <1–6% (~7 mm Hg–43 mm Hg), as compared to most normal tissues of 2% to 9% (14–65 mm Hg) [87]. This hypoxic, acidic microenvironment generates a redox imbalance, which, combined with a slightly hypertonic medium and TGF-β and BMP signaling, is sufficient to drive cancer cells to a gene expression pattern with characteristic features of the dormancy signature [147].
In the osteoblast niche, data suggest that metastatic malignant cells usurp the HSC niche to create an abnormal niche that is unable to support normal hematopoiesis [63,132,136,148]. BC cells with a stem cell phenotype compete with HSCs in the endosteal niche and remain dormant in a Notch-dependent manner by spindle-shaped N-cadherin+ CD45− osteoblasts (SNO cells) [149]. Micrometastases survive in the HSC niche close to endosteal stromal cells [132], where preosteoblasts support their survival and chemoresistance [9], partially mediated by Jagged1 [150]. As noted, it is the preosteoblasts that likely support dormancy [48], but as osteoblasts differentiate, they connect with cancer cells through gap junctions, increase their intracellular calcium levels and potentially promote colonization [53]. Jagged-1/Notch signaling regulates tumor stem cell development, epithelial-to-mesenchymal transition, and immune cell homeostasis during minimal residual disease, and plays a role in the recurrence of minimal residual disease in primary tumors [151]. However, some osteoblasts in the HSC niche become “educated” by arriving cancer cells to support the dormant state [152]. These “educated” osteoblasts express RUNX2/osteocalcin (OCN)/OPN, are negative for IL-6 and α-smooth muscle actin (αSMA), and have new properties where they acquire the capacity to suppress both triple-negative and ER+ breast cancer cell proliferation [153]. They increase cancer cell p21Waf1 expression [152], regulate ERK 1 and 2 signaling and inhibit S-phase entry [153]. These effects are mediated by the secretion of small extracellular vesicles enriched for miR-148a-3p [153]. These data underscore the reciprocal relationship between the cancer cells educating the metastatic microenvironment in the premetastatic and dormancy niches, and the dormancy-endowing effects of the niche on cancer cells.
In the vascular niche, non-sprouting endothelial cells produce ECM components such as thrombospondin-1 (TSP1), which may induce dormancy [46]. The endothelial Duffy antigen receptor for chemokines (DARC) may induce dormancy in cancer cells by binding to the metastasis suppressor cluster of differentiation 82 (KAI1), inhibiting proliferation through p21Waf1 and downregulating T-Box transcription factor 2 (TBX2) [154]. Signaling mechanisms associated with micrometastasis dormancy include von Willebrand factor (vWF) [8], VCAM1 [8], CXCL 1 and 2 [155], BMP7 [90], TGFβ-2 [91], canonical nuclear factor (NF)κB combined with ER signaling in ER+ BC cells [156], nuclear receptor subfamily 2 group F member (NR2F)1 [93] and zing finger protein (ZFP)281 [157]. Dormant stem cell signaling through phosphatase and tensin homolog (PTEN) maintains a dominant role in tumorigenic stimuli [92]. Perivascular periaxin (Prx)1+ MSCs express CXCL12 and maintain quiescence and chemoresistance of leukemic stem cells, in contrast to their effects on HSCs, suggesting a more complex mechanism that differentiates the roles of the endothelial niche in malignant vs. normal hematopoietic stem cell maintenance [158].
However, signaling in the vascular endothelial niche is not all dormancy-inducing in malignant cells [159]. Analogous to the case of hematopoietic stem cells that receive pro-differentiating signals once they translocate to the endothelial niche, cancer cell micrometastases can receive context-specific proliferative signals in their interactions with endothelial cell tips mediated through TGF-β1 and periostin [159]. Indeed, the effects of periostin appear to be context-specific, as some of its effects on HSC are linked to stem cell maintenance in the endosteal niche, as noted above [54]. Endothelial cells can promote a stem-like phenotype in some solid tumor cancer cells through the activation of the hedgehog pathway through Gli-1 [160]. Gli-1 expression is high in breast cancer and contributes to therapeutic resistance in both ER+ [161] and ER− BC cells [162,163] through Wnt signaling [162]. Endothelial cell L1 cell adhesion molecule (L1CAM) ligands may induce the proliferation of L1CAM+ DTCs [94]. Endothelial leukocyte adhesion molecule 1 (E-selectin) signaling in endothelial cells induces a non-canonical mesenchymal–epithelial transition (MET) phenotype in cancer cells, which begin to express EpCam and cytokeratin 14 (CK14) while continuing to express mesenchymal gene expression factors including snail family transcriptional repressor (Snail) 1/2, twist family bHLH transcription factor (Twist) 1/2, zinc finger E-box binding homeobox (Zeb) 1/2 and cancer stem cell marker Sox 2/9 [164]. These programs permit the regrowth of dormant micrometastases [164]. The conditions for the recurrence of cells expressing mesenchymal programs are discussed below [121]. Quiescent DTCs in the BM lack the epithelial marker E-cadherin [165], but do not undergo a phenotypic appearance of EMT [166]. This is corroborated by in vitro data supporting a model for continued mesenchymal signaling in dormant cells with an apparent epithelial phenotype [120,121], as discussed below. However, once these cells are stimulated to undergo MET, they begin to proliferate once again. Micrometastatic sites can serve as launching pads for colonization and re-metastasis [167].
The immune niche contains macrophages and CD4+ and CD8+ cells that may induce dormancy [166] through INFγ [46]. Quiescent cancer cells in distant organs that have tumor-initiating capacity express DKK-1, which inhibits Wnt, enhancing the downregulation of Natural Killer (NK) cell activators and death ligands, and evading killing by NK cells [168]. In addition to homing, SDF-1/CXCR4 may promote survival through Src through Akt and TNF resistance through TNF-related apoptosis-inducing ligand (TRAIL) [169]. Secretion of SDF-1α by BM MSCs may maintain quiescence in breast cancer micrometastases by downregulating the truncated neurokinin receptor-1 (NK1R-Tr) expression [128].
Signaling intrinsic to the cancer cell is also likely to contribute to the dormant state by expressing metastasis suppressor genes that contribute to dormancy without affecting the growth of cells in the originating primary tumor [170]. The tyrosine kinase receptor TIE2, which induces dormancy in hematopoietic stem cells, also induces cell cycle arrest in breast cancer cells through CDK inhibitors CDKN1A (p21Waf1) and CDKN1B (p27Kip1) in vitro, decreases osteolytic metastases and response to antimetabolites in mice, and is associated with delayed time to metastasis in breast cancer patients [171]. Expression of the metastasis suppressor genes KISS-1, metastasis suppressor Kangai-1 (KAI1), mitogen-activated protein kinase (MKK)4/7 and NM23 nucleoside diphosphate kinase 1 (Nm23-H1) by cancer cells also promotes tumor dormancy at the metastatic site [170]. Signaling through indoleamine 2,3-dioxygenase 1 (IDO1) through the mammalian target of rapamycin (mTOR) and control of nonderepressible-2 kinase has been linked to cellular quiescence [172]. IDO1, which is a heme-containing enzyme that mediates the rate-limiting step in the metabolism of l-tryptophan to kynurenine, has been explored as a potential immunotherapeutic target in oncology [173]. An inhibitor of this pathway has been found to have an acceptable toxicological spectrum in animal studies [173].
Other mechanisms of inducing reversible dormancy functions through epigenetic modifications, such as repressive histones [174] or the downregulation of suppressor gene promoter methylation enzymes [175], have been explored. These effects are analogous to evolutionary mechanisms that ensure the survival of organisms in environmentally disadvantageous circumstances [176]. They can also originate in the primary tumor, where epigenetic modifications in some of the cells enable them to enter dormancy in a distant microenvironment by cancer-associated fibroblasts (CAF) with altered p53 functions [177]. In tumor cells, the downregulation of DNA methyltransferase (DNMT)1 expression results in silencing a transcription network regulating the G1-S transition, including forkhead box (FOX)M1, FOXD, FOXL, early growth response (EGR)1/2/3, peroxisome proliferator-activated receptor (PPAR)γ, ETS Like-1 protein Elk-1 (ELK1) and Jun family members [78,176]. However, the dormancy-associated genes p53, DEC2, nuclear receptor subfamily 2 group F member (NR2F)1 and retinoic acid receptor (RAR)β, which are often silenced in proliferating cancer, are upregulated in dormant cells [78,176]. NR2F1 and RARβ together direct the removal of acetyl groups from histone H3 by histone deacetylases (HDACs) and are associated with dormant DTCs in patients [20,93,178]. In contrast, NR2F1 induces the methylation of H3 residues histone H3 (H3)K4, H3K9 and H3K27 and decreases the expression of growth-promoting SOX9 [176]. Histone H4 methylation is necessary for breast cancer dormancy in the lungs [179]. Epigenetics also affect dormancy and proliferation by governing the processing of coding mRNA alternative isoforms and non-coding RNAs, including micro-RNAs and long non-coding RNAs [180,181].
The overall effect of the metastatic microenvironment is to impose a reversible state of dormancy on the microscopic disseminated tumor-initiating cells. This effect is mediated through cancer cell interactions with structural, soluble, cellular and biophysical elements of the microenvironment that initiate signaling through a variety of receptors and sensors outlined above in order to change gene expression and phenotypic patterns to induce a dormant, cytotoxin-resistant state.
Table 1. Mechanisms of breast cancer dormancy in the bone marrow.
| Vehicle | Signaling | References | |
|---|---|---|---|
| Endosteal niche |
MSCs |
MERTK, AXL, TGFβR3, BMPR2, Alk5, NDRG1, ERK, p38, p21WAF1, p27Kip1, 15INK4b, PI3K, RhoA/GRAF, integrin α5β1, FGF-2, HSPG, fibronectin | [36,46,81,88,90,91,93,94,98,99,100,102,103,105,114,115,119,120,121,122,123,124,125,126,127,128,129,130,131,132,133,134,135,136] |
| Inhibition of oncogene signaling | [92] | ||
| Non-canonical Wnt5a signaling, SIAH2, repression of β-catenin, LIF, RA | [94,142,143] | ||
| Hypoxia, acidic pH | LIF, STAT3, TGFβ, BMP signaling | [87,139,147] | |
| Redox signaling | [140] | ||
| Exosomes | miR-23b, -126, 127, -148a, -3p -197, -222, -223 | [94,95,125,126,127,141,153] | |
| Fusion with and cannibalizing MSCs | SDF-1a, decreased CXCL12 | [126,128,144,146] | |
| Microenvironmental remodeling | [129] | ||
| Preosteoblasts, SNO cells | Notch, Jagged1 | [48,149,150] | |
| Vascular niche | Endothelial cells | TSP1 | [46] |
| DARC, KAI1, p21Waf1, downregulated TBX2 |
[154] | ||
| vWF, VCAM1, CXCL 1 and 2, BMP7, TGFβ-2, NFκB combined with ER in ER+ BC, NR2F1, ZFP281, PTEN | [8,91,92,93,155,156,157] |
||
| Prx1+ MSCs | CXCL12 | [158] | |
| Immune niche | CD4+ and CD8+ cells | INFγ | [46,166] |
| NK cells | DKK-1, inhibited canonical Wnt | [168] | |
| SDF-1/CXCR4 | Src, Akt, TRAIL, downregulated NK1R-Tr | [128,169] | |
| Cancer cell-intrinsic effects | TIE2 | p21Waf1, p27Kip1 | [170,171] |
| KAI1, MKK4/7, Nm23-H1 | [170] | ||
| IDO1 | mTOR | [172] | |
| Epigenetics | Repressive histones | altered p53 functions | [174,175,177] |
| downregulation of suppressor gene promoter methylation enzymes | |||
| downregulation of DNMT1 |
silencing of a transcription network FOXM1, FOXD, FOXL EGR1/2/3, PPARγ, ELK1, Jun family upregulating p53, DEC2, NR2F1, RARβ |
[78,176] |
|
| NR2F1, RARβ | removal of acetyl groups from histone H3, HDACs | [20,93,178] | |
| NR2F1 |
induced methylation of H3 residues H3K4, H3K9, H3K27, decreased expression of growth-promoting SOX9 | [176] |
|
| processing alternative coding mRNA isoforms, non-coding RNAs, miRNAs, lnRNAs | [180,181] |
This entry is adapted from the peer-reviewed paper 10.3390/cancers15113021
This entry is offline, you can click here to edit this entry!