The 16S rRNA gene is highly conserved in all bacteria (and also archaea). Nonetheless, it contains nine hypervariable regions (V1 - V9), where sequences of these regions can be used to identify and discriminate bacterial genus, sometimes until the species level. This makes the gene a useful tool for phylogenetic studies. With the introduction of next-generation sequencing technologies, 16S rRNA next-generation sequencing (16SNGS) has allowed profiling of bacterial communities found in organisms and the environment, and lead to the discovery of many previously unculturable members of the bacteria kingdom.
- 16S rRNA
- bacterial identification
- 16S rRNA next-generation sequencing workflow
1. Introduction
With the advent of Sanger DNA sequencing, bacteria can now be identified via nucleotide sequence of the 16S rRNA gene—a short, conserved gene specific to bacterial genus (96%) and for some, species (87.5%)[1]. This method of bacterial identification is culture-independent and only requires DNA of the tested bacteria[2]. The development of NGS techniques, including 16S rRNA next-generation sequencing (16SNGS), allows further upscaling of sequencing quantity (fragments versus time) even in mixed cultures.
2.16SNGS: Platforms, Workflow and Bioinformatics Analysis
2.1. NGS Technology
The era of NGS began with the introduction of pyrosequencing technology by 454 Life Sciences in 2005, followed by the Solexa/Illumina platform, Life Technologies’ SOLiD, Ion Torrent and Ion Proton sequencers, and later, the MiSeq and HiSeq platforms from Illumina[3][4][5]. Even though pyrosequencing is considered the “pioneer” of NGS, it is now no longer available after the platform was discontinued by Roche in 2015. The Illumina and Ion fleet of sequencers operate using a “sequencing by synthesis” chemistry[6], compared to the “sequencing by ligation” technology of the now (also) discontinued SOLiD platform[7]. Due to its ability to read palindromic sequences, “sequencing by synthesis” appears to be the more popular chemistry[8], resulting in the dominance of both Illumina and Ion sequencers (currently: Ion Torrent Genexus System and Ion Gene Studio S5 System) in NGS laboratories. Illumina sequencers are now available in either benchtop or production scale categories, allowing users more options to select the best NGS platform according to their laboratory needs and budget allocations (https://www.illumina.com/systems/sequencing-platforms.html). Of note, the most recent player in the NGS industry will be Complete Genomics (acquired by Beijing Genome Institute (BGI))’s “DNA nanoballs” sequencing platforms (https://en.mgitech.cn/products/)[9]; these platforms were reported to be comparable in performance to Illumina sequencers[10][11][12]. In the recent decade, third-generation or long-read sequencing technologies by Pacific Biosciences and Oxford Nanopore Technology have also been launched[13][14][15], although, due to cost concerns, these platforms are mostly used in research institutes compared to diagnostic laboratories. For a comprehensive overview of NGS platforms and their associated chemistries, readers may refer to reviews by Ambardar et al.[16], Besser et al.[17] and Slatko et al. [18]. Regardless of their sequencing chemistry, all NGS platforms can generate millions of DNA molecules with different yields and sequence lengths via parallel sequencing, and allow simultaneous multiplexing of several hundred samples in a single run[19].
2.2. 16SNGS Workflow and Bioinformatics Analysis
Introduction of NGS technologies significantly promoted the development of “metagenomics”. The term “metagenomics” was first used by Handelsman et al. over 20 years ago, where it refers to the study of genetic material from a sample without the need for isolation and culturing of the microorganisms contained in the sample itself[20][21]. Originally used to study microbial community diversity within samples from the environment and also organisms, laboratories built on the metagenome concept to sequence only the 16S rRNA gene of bacterial populations within specimens to identify bacteria[4][22]. Nevertheless, the workflow for bacteria identification via 16SNGS is vastly different from that of the conventional protocol of culture and biochemical testing (CBtest) (Figure 1).
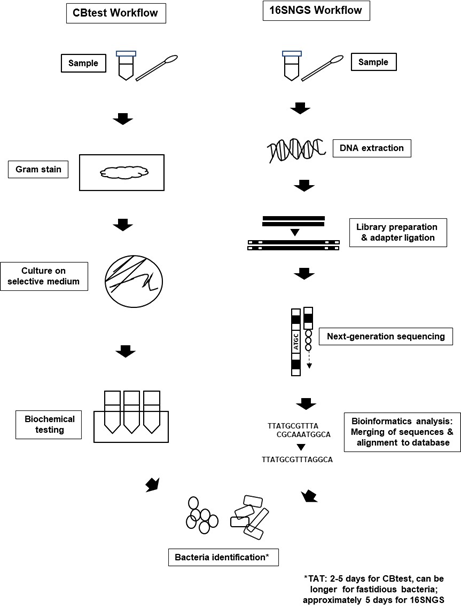
Figure 1. Bacterial identification workflow via CBtest is different from that of 16SNGS. For CBtest, samples are subjected to gram staining and culture on selective medium. Subsequent biochemical testing will reveal the identity of the bacteria. On the other hand, DNA extraction from samples is carried out in the first step of 16SNGS workflow. After library preparation, NGS of the 16S rRNA fragments will then be done, followed by bioinformatics analysis to identify the bacterial sample. Turn-around-time (TAT) of the identification process is workflow-dependent.
Briefly, the 16SNGS workflow starts with genomic DNA extraction of bacteria from collected samples. Genomic DNA is extracted using either conventional protocols or commercialized kits, and subsequently quantified to determine quantity and quality of the extracted DNA[23][24]. Following this, 16S rRNA gene libraries are prepared, from which variable regions of the 16S rRNA gene will be amplified[5][25]. Depending on the sequencing platform, the variable region selected for amplification and sequence for bacterial identification may differ, though it has been reported that the V4-V6 regions are most representative of the full-length 16S rRNA gene[26][27][28][29] . Subsequently, DNA pre-processing to obtain specific sizes of DNA fragments is carried out. Adapters will then be added onto the amplified 16S rRNA region. Following this, quantification and normalization of the amplicons will be carried out prior to sequencing[5][30][31][32].
Increasingly, the processes of DNA extraction and library preparation have been identified as potential bottlenecks of the NGS workflow, especially in a diagnostic laboratory dealing with a large number of samples, daily. To counter this, automated nucleic acid extraction machines, such as the QIAcube (Qiagen Inc.), Maxwell® RSC (Promega Corporation) and KingFisher automated extraction and purification platforms (Thermo Fisher Scientific) allow walk-away DNA extraction[33], and are now an essential component in many large NGS centers. In addition, automation in liquid handling for library preparation is achievable via pipetting workstations such as the Biomek i-Series (Beckman Coulter and Bravo Automated Liquid Handling Platform (Agilent)[34]. In future, microfluidics solutions for NGS library preparation will enable miniaturization and enclosed environment for the process, minimizing contamination and optimizing laboratory space utilization[35][36][37].
After sequencing is completed, raw data pre-processing is important prior to bioinformatics analysis. This includes the screening and removal of sequencing adapters, assessing overall sequencing read quality (quality checking), trimming or filtering low quality reads and filtering of reads based on sequence length. This step is important for removing low quality and erroneous reads. To detect putative chimeric sequences in filtered data, the sequences are normally subjected to chimera check[38]. At this stage, all chimeric sequences are removed before the next step of analysis. For data pre-processing, multiple tools are freely available, including PEAT[39], Trimmomatic [40] and FastQC (Babraham Bioinformatics, Cambridge). For paired-end data, the merging of forward and reverse reads is done as the first step of quality control and could be performed with BBMerge[41]. To analyze the 16S rRNA gene in bacteria, a common approach is via operational taxonomic unit (OTU) clustering, where sequences are clustered into a representative OTU sequence, defined at ≥97% sequence similarity level[42][43]. The OTU-based approach is used to distinguish and differentiate biologically real nucleotide differences from artefacts[44]. The primary output of this approach will be OTU tables represented by BIOM file format. Quantitative Insight into Microbial Ecology (QIIME)[45] is one of the most popular tools for the OTU-based approach. Recently, the amplicon sequence variants (ASVs) approach has been introduced; several pipelines are now available with the aim to correct sequencing errors and improve taxonomic resolution, including DADA2[46], Qiime2-Deblur[47] and USEARCH-UNOISE3[48]. Sensitivity and specificity differ between different pipelines, among which DADA2 was reported to have the best sensitivity and resolution. Even though it produces a higher number of spurious ASV compared to others, DADA2 would still be the best choice to obtain the highest possible resolution[49].
Subsequent alignment of the consensus sequences to a reference database will identify the bacteria being investigated. Public repositories of bacterial 16S rRNA gene database are available for this purpose. The NCBI Bacterial 16S Ribosomal RNA RefSeq Targeted Loci Project (https://www.ncbi.nlm.nih.gov/bioproject/33175) curates comprehensive and non-redundant 16S rRNA sequences submitted by the public to the International Nucleotide Sequence Database Collaboration (INSDC)[50]. On the other hand, the Ribosomal Database project hosted by Michigan State University[51] also contains 16S rRNA sequences from INSDC, though it has a smaller source of taxonomy classification compared to NCBI[52]. Pathogen identification can also be done using SILVA (https://www.arb-silva.de), which provides aligned rDNA sequences from Bacteria, Archaea and Eukaryota domains [53].
The Greengenes database (http://greengenes.secondgenome.com) is the default database in the QIIME pipeline. Nonetheless, this database is one of the more popular database used in 16SNGS[54]. However, the database has not been updated since 2014, and may not contain nomenclature of novel or renamed bacteria after 2014. In recent years, new 16S rRNA gene databases such as the GRD—Genomic-based 16S ribosomal RNA Database (https://metasystems.riken.jp/grd/) and the EzBioCloud 16S database (https://www.ezbiocloud.net/resources/16s_download) have been established. GRD curators correct misannotations or missing anti-SD sites and other short segments of the 16S rRNA gene sequences extracted from complete genomes for more reliable taxonomic assignments. EzBioCloud 16S database is a commercial product; nevertheless, at the time of writing, it is freely available for users from academic and non-profit institutions. The database has been shown to allow bacterial identification to species level and provided taxonomic accuracy comparable to SILVA and Greengenes [55].
This entry is adapted from the peer-reviewed paper 10.3390/diagnostics10100816
References
- Srinivasan, R.; Karaoz, U.; Volegova, M.; MacKichan, J.; Kato-Maeda, M.; Miller, S.; Nadarajan, R.; Brodie, E.L.; Lynch, S.V. Use of 16S rRNA gene for identification of a broad range of clinically relevant bacterial pathogens. PLoS ONE 2015, 10, e0117617, doi:10.1371/journal.pone.0117617.
- Sune, D.; Rydberg, H.; Augustinsson, Å.N.; Serrander, L.; Jungeström, M.B. Optimization of 16S rRNA gene analysis for use in the diagnostic clinical microbiology service. J. Microbiol. Methods 2020, 170, 105854, doi:10.1016/j.mimet.2020.105854.
- Kwong, J.C.; Mccallum, N.; Sintchenko, V.; Howden, B.P. Whole genome sequencing in clinical and public health microbiology. Pathology 2015, 47, 199–210, doi:10.1097/PAT.0000000000000235.
- Escobar-Zepeda, A.; Vera-Ponce de León, A.; Sanchez-Flores, A. The Road to Metagenomics: From Microbiology to DNA Sequencing Technologies and Bioinformatics. Front. Genet. 2015, 6, 348, doi:10.3389/fgene.2015.00348.
- Buermans, H.P.J.; Den Dunnen, J.T. Next generation sequencing technology: Advances and applications. Biochim. Biophys. Acta Mol. Basis Dis. 2014, 1842, 1932–1941, doi:10.1016/j.bbadis.2014.06.015.
- Meyer, M.; Kircher, M. Illumina sequencing library preparation for highly multiplexed target capture and sequencing. Cold Spring Harb. Protoc. 2010, 2010, pdb.prot5448, doi:10.1101/pdb.prot5448.
- Valouev, A.; Ichikawa, J.; Tonthat, T.; Stuart, J.; Ranade, S.; Peckham, H.; Zeng, K.; Malek, J.A.; Costa, G.; McKernan, K.; et al. A high-resolution, nucleosome position map of C. elegans reveals a lack of universal sequence-dictated positioning. Genome Res. 2008, 18, 1051–1063, doi:10.1101/gr.076463.108.
- Huang, Y.-F.; Chen, S.-C.; Chiang, Y.-S.; Chen, T.-H.; Chiu, K.-P. Palindromic sequence impedes sequencing-by-ligation mechanism. BMC Syst. Biol. 2012, 6 (Suppl. 2), S10, doi:10.1186/1752-0509-6-S2-S10.
- Porreca, G.J. Genome sequencing on nanoballs. Nat. Biotechnol. 2010, 28, 43–44.
- Patch, A.-M.; Nones, K.; Kazakoff, S.H.; Newell, F.; Wood, S.; Leonard, C.; Holmes, O.; Xu, Q.; Addala, V.; Creaney, J.; et al. Germline and somatic variant identification using BGISEQ-500 and HiSeq X Ten whole genome sequencing. PLoS ONE 2018, 13, e0190264.
- Mak, S.S.T.; Gopalakrishnan, S.; Carøe, C.; Geng, C.; Liu, S.; Sinding, M.-H.S.; Kuderna, L.F.K.; Zhang, W.; Fu, S.; Vieira, F.G.; et al. Comparative performance of the BGISEQ-500 vs Illumina HiSeq2500 sequencing platforms for palaeogenomic sequencing. Gigascience 2017, 6, 1–13, doi:10.1093/gigascience/gix049.
- Zhu, F.-Y.; Chen, M.-X.; Ye, N.-H.; Qiao, W.-M.; Gao, B.; Law, W.-K.; Tian, Y.; Zhang, D.; Zhang, D.; Liu, T.-Y.; et al. Comparative performance of the BGISEQ-500 and Illumina HiSeq4000 sequencing platforms for transcriptome analysis in plants. Plant. Methods 2018, 14, 69, doi:10.1186/s13007-018-0337-0.
- Eid, J.; Fehr, A.; Gray, J.; Luong, K.; Lyle, J.; Otto, G.; Peluso, P.; Rank, D.; Baybayan, P.; Bettman, B.; et al. Real-time DNA sequencing from single polymerase molecules. Science 2009, 323, 133–138, doi:10.1126/science.1162986.
- Kai, S.; Matsuo, Y.; Nakagawa, S.; Kryukov, K.; Matsukawa, S.; Tanaka, H.; Iwai, T.; Imanishi, T.; Hirota, K. Rapid bacterial identification by direct PCR amplification of 16S rRNA genes using the MinIONTM nanopore sequencer. FEBS Open Bio 2019, 9, 548–557, doi:10.1002/2211-5463.12590.
- Schloss, P.D.; Jenior, M.L.; Koumpouras, C.C.; Westcott, S.L.; Highlander, S.K. Sequencing 16S rRNA gene fragments using the PacBio SMRT DNA sequencing system. PeerJ 2016, 4, e1869, doi:10.7717/peerj.1869.
- Ambardar, S.; Gupta, R.; Trakroo, D.; Lal, R.; Vakhlu, J. High Throughput Sequencing: An Overview of Sequencing Chemistry. Indian J. Microbiol. 2016, 56, 394–404, doi:10.1007/s12088-016-0606-4.
- Besser, J.; Carleton, H.A.; Gerner-Smidt, P.; Lindsey, R.L.; Trees, E. Next-generation sequencing technologies and their application to the study and control of bacterial infections. Clin. Microbiol. Infect. 2018, 24, 335–341, doi:10.1016/j.cmi.2017.10.013.
- Slatko, B.E.; Gardner, A.F.; Ausubel, F.M. Overview of Next-Generation Sequencing Technologies. Curr. Protoc. Mol. Biol. 2018, 122, e59, doi:10.1002/cpmb.59.
- Alekseyev, Y.O.; Fazeli, R.; Yang, S.; Basran, R.; Maher, T.; Miller, N.S.; Remick, D. A Next-Generation Sequencing Primer-How Does It Work and What Can It Do? Acad. Pathol. 2018, 5, 2374289518766521, doi:10.1177/2374289518766521.
- Handelsman, J.; Rondon, M.; Brady, S.; Clardy, J.; Goodman, R.; Handelsman, J.; Rondon, M.R.; Brady, S.F.; Clardy, J.; Goodman, R.M. Molecular Biological access to the chemistry of unknown soil microbes: A new frontier for natural products. Chem Biol 5: R245-R249. Chem. Biol. 1998, 5, R245–R249, doi:10.1016/S1074-5521(98)90108-9.
- Chen, K.; Pachter, L. Bioinformatics for Whole-Genome Shotgun Sequencing of Microbial Communities. PLOS Comput. Biol. 2005, 1, e24.
- Deurenberg, R.H.; Bathoorn, E.; Chlebowicz, M.A.; Couto, N.; Ferdous, M.; García-Cobos, S.; Kooistra-Smid, A.M.D.; Raangs, E.C.; Rosema, S.; Veloo, A.C.M.; et al. Application of next generation sequencing in clinical microbiology and infection prevention. J. Biotechnol. 2017, 243, 16–24, doi:10.1016/j.jbiotec.2016.12.022.
- Watanabe, N.; Kryukov, K.; Nakagawa, S.; Takeuchi, J.S.; Takeshita, M.; Kirimura, Y.; Mitsuhashi, S.; Ishihara, T.; Aoki, H.; Inokuchi, S.; et al. Detection of pathogenic bacteria in the blood from sepsis patients using 16S rRNA gene amplicon sequencing analysis. PLoS ONE 2018, 13, e0202049.
- Sabat, A.J.; Van Zanten, E.; Akkerboom, V.; Wisselink, G.; Van Slochteren, K.; De Boer, R.F.; Hendrix, R.; Friedrich, A.W.; Rossen, J.W.A.; Kooistra-Smid, A.M.D. (Mirjam) Targeted next-generation sequencing of the 16S-23S rRNA region for culture-independent bacterial identification–increased discrimination of closely related species. Sci. Rep. 2017, 7, 3434, doi:10.1038/s41598-017-03458-6.
- Bartram, A.K.; Lynch, M.D.J.; Stearns, J.C.; Moreno-Hagelsieb, G.; Neufeld, J.D. Generation of Multimillion-Sequence 16S rRNA Gene Libraries from Complex Microbial Communities by Assembling Paired-End Illumina Reads. Appl. Environ. Microbiol. 2011, 77, 3846–3852, doi:10.1128/AEM.02772-10.
- Watts, G.S.; Youens-Clark, K.; Slepian, M.J.; Wolk, D.M.; Oshiro, M.M.; Metzger, G.S.; Dhingra, D.; Cranmer, L.D.; Hurwitz, B.L. 16S rRNA gene sequencing on a benchtop sequencer: Accuracy for identification of clinically important bacteria. J. Appl. Microbiol. 2017, 123, 1584–1596, doi:10.1111/jam.13590.
- Bukin, Y.S.; Galachyants, Y.P.; Morozov, I.V.; Bukin, S.V.; Zakharenko, A.S.; Zemskaya, T.I. The effect of 16S rRNA region choice on bacterial community metabarcoding results. Sci. Data 2019, 6, 190007, doi:10.1038/sdata.2019.7.
- Fouhy, F.; Clooney, A.G.; Stanton, C.; Claesson, M.J.; Cotter, P.D. 16S rRNA gene sequencing of mock microbial populations- impact of DNA extraction method, primer choice and sequencing platform. BMC Microbiol. 2016, 16, 123, doi:10.1186/s12866-016-0738-z.
- Yang, B.; Wang, Y.; Qian, P.Y. Sensitivity and correlation of hypervariable regions in 16S rRNA genes in phylogenetic analysis. BMC Bioinform. 2016, 17, 1–8, doi:10.1186/s12859-016-0992-y.
- Inc., Illumina. Part # 15044223 Rev. B. In 16S Metagenomic Sequencing Library Preparation; Illumina Inc.: San Diego, CA, USA, 2013.
- Thermo Fisher Scientific. MAN0010799. In Ion. 16S Metanomics Kit; Thermo Fisher Scientific: Waltham, MA, USA, 2020.
- Pichler, M.; Coskun, Ö.K.; Ortega-Arbulú, A.-S.; Conci, N.; Wörheide, G.; Vargas, S.; Orsi, W.D. A 16S rRNA gene sequencing and analysis protocol for the Illumina MiniSeq platform. Microbiologyopen 2018, 7, e00611, doi:10.1002/mbo3.611.
- Tan, S.C.; Yiap, B.C. DNA, RNA, and protein extraction: The past and the present. J. Biomed. Biotechnol. 2009, 2009, 574398, doi:10.1155/2009/574398.
- Hess, J.F.; Kohl, T.A.; Kotrová, M.; Rönsch, K.; Paprotka, T.; Mohr, V.; Hutzenlaub, T.; Brüggemann, M.; Zengerle, R.; Niemann, S.; et al. Library preparation for next generation sequencing: A review of automation strategies. Biotechnol. Adv. 2020, 41, 107537, doi:10.1016/j.biotechadv.2020.107537.
- Kim, H.; Jebrail, M.J.; Sinha, A.; Bent, Z.W.; Solberg, O.D.; Williams, K.P.; Langevin, S.A.; Renzi, R.F.; Van De Vreugde, J.L.; Meagher, R.J.; et al. A Microfluidic DNA Library Preparation Platform for Next-Generation Sequencing. PLoS ONE 2013, 8, e68988.
- Tan, S.J.; Phan, H.; Gerry, B.M.; Kuhn, A.; Hong, L.Z.; Min Ong, Y.; Poon, P.S.Y.; Unger, M.A.; Jones, R.C.; Quake, S.R.; et al. A Microfluidic Device for Preparing Next Generation DNA Sequencing Libraries and for Automating Other Laboratory Protocols That Require One or More Column Chromatography Steps. PLoS ONE 2013, 8, e64084.
- Hess, J.F.; Kotrová, M.; Calabrese, S.; Darzentas, N.; Hutzenlaub, T.; Zengerle, R.; Brüggemann, M.; Paust, N. Automation of Amplicon-Based Library Preparation for Next-Generation Sequencing by Centrifugal Microfluidics. Anal. Chem. 2020, doi:10.1021/acs.analchem.0c01202.
- Cole, J.R.; Wang, Q.; Fish, J.A.; Chai, B.; McGarrell, D.M.; Sun, Y.; Brown, C.T.; Porras-Alfaro, A.; Kuske, C.R.; Tiedje, J.M. Ribosomal Database Project: Data and tools for high throughput rRNA analysis. Nucleic Acids Res. 2014, 42, D633–D642, doi:10.1093/nar/gkt1244.
- Li, Y.-L.; Weng, J.-C.; Hsiao, C.-C.; Chou, M.-T.; Tseng, C.-W.; Hung, J.-H. PEAT: An intelligent and efficient paired-end sequencing adapter trimming algorithm. BMC Bioinform. 2015, 16 (Suppl. 1), S2, doi:10.1186/1471-2105-16-S1-S2.
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120, doi:10.1093/bioinformatics/btu170.
- Bushnell, B.; Rood, J.; Singer, E. BBMerge—Accurate paired shotgun read merging via overlap. PLoS ONE 2017, 12, e0185056, doi:10.1371/journal.pone.0185056.
- Nguyen, N.-P.; Warnow, T.; Pop, M.; White, B. A perspective on 16S rRNA operational taxonomic unit clustering using sequence similarity. NPJ Biofilms Microbiomes 2016, 2, 16004, doi:10.1038/npjbiofilms.2016.4.
- Bharti, R.; Grimm, D.G. Current challenges and best-practice protocols for microbiome analysis. Brief. Bioinform. 2019, doi:10.1093/bib/bbz155.
- Nearing, J.T.; Douglas, G.M.; Comeau, A.M.; Langille, M.G.I. Denoising the Denoisers: An independent evaluation of microbiome sequence error-correction approaches. PeerJ 2018, 6, e5364, doi:10.7717/peerj.5364.
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336, doi:10.1038/nmeth.f.303.
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583, doi:10.1038/nmeth.3869.
- Amir, A.; McDonald, D.; Navas-Molina, J.A.; Kopylova, E.; Morton, J.T.; Zech Xu, Z.; Kightley, E.P.; Thompson, L.R.; Hyde, E.R.; Gonzalez, A.; et al. Deblur Rapidly Resolves Single-Nucleotide Community Sequence Patterns. mSystems 2017, 2, doi:10.1128/mSystems.00191-16.
- Edgar, R.C. UNOISE2: Improved error-correction for Illumina 16S and ITS amplicon sequencing. bioRxiv 2016, 81257, doi:10.1101/081257.
- Prodan, A.; Tremaroli, V.; Brolin, H.; Zwinderman, A.H.; Nieuwdorp, M.; Levin, E. Comparing bioinformatic pipelines for microbial 16S rRNA amplicon sequencing. PLoS ONE 2020, 15, e0227434.
- Federhen, S. The NCBI Taxonomy database. Nucleic Acids Res. 2011, 40, D136–D143, doi:10.1093/nar/gkr1178.
- Cole, J.R.; Wang, Q.; Fish, J.A.; Chai, B.; McGarrell, D.M.; Sun, Y.; Brown, C.T.; Porras-Alfaro, A.; Kuske, C.R.; Tiedje, J.M. Ribosomal Database Project: Data and tools for high throughput rRNA analysis. Nucleic Acids Res. 2014, 42, D633–D642, doi:10.1093/nar/gkt1244
- Balvočiūtė, M.; Huson, D.H. SILVA, RDP, Greengenes, NCBI and OTT—How do these taxonomies compare? BMC Genom. 2017, 18, 114, doi:10.1186/s12864-017-3501-4.
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2012, 41, D590–D596, doi:10.1093/nar/gks1219.
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a Chimera-Checked 16S rRNA Gene Database and Workbench Compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072, doi:10.1128/AEM.03006-05.
- Park, S.-C.; Won, S. Evaluation of 16S rRNA Databases for Taxonomic Assignments Using Mock Community. Genom. Inform. 2018, 16, e24, doi:10.5808/GI.2018.16.4.e24.
- Cole, J.R.; Wang, Q.; Fish, J.A.; Chai, B.; McGarrell, D.M.; Sun, Y.; Brown, C.T.; Porras-Alfaro, A.; Kuske, C.R.; Tiedje, J.M. Ribosomal Database Project: Data and tools for high throughput rRNA analysis. Nucleic Acids Res. 2014, 42, D633–D642, doi:10.1093/nar/gkt1244