Since the discovery of insulin 100 years ago, a primary focus of insulin research has been to understand how this metabolic hormone regulates glucose homeostasis. The body of evidence gained has been fundamental for developing treatment strategies for conditions involving insulin resistance, including obesity-sensitive type 2 diabetes [
1]. However, the actions of insulin in the body are not just metabolic in nature, and are not restricted to the periphery. Pancreatic insulin can enter the brain slowly at the level of the choroid plexus, which produces cerebrospinal fluid (CSF) in the ventricles of the brain. Ventricular CSF is contiguous with brain interstitial fluid, allowing a gradual increase in brain insulin levels following insulin release in the periphery. A more direct and rapid source for brain entry, however, is by trans-endothelial transport at the level of brain microvasculature, i.e., by crossing the blood–brain barrier (BBB) [
2,
3,
4,
5]. In addition, it has been suggested that insulin may be produced and released at the choroid plexus [
6], and even by some neurons [
7,
8] and astrocytes [
9] within the brain, providing an even more local and rapid source of insulin.
Physiological concentrations of insulin in CSF and the brain are estimated to reach 10 to 30 nM after food intake [
2,
10,
11,
12,
13]. Within the brain, mRNA transcripts and protein for insulin receptors (InsRs) are distributed widely with high levels in the olfactory bulb, hypothalamus, hippocampus, cortex, cerebellum, midbrain and striatum [
14,
15,
16,
17,
18,
19,
20]. Thus, insulin has the potential to impact multiple brain regions and their functions through InsR signaling. Insulin also acts at insulin-like growth factor 1 receptors (IGF-1Rs), albeit at higher concentrations than those required to activate InsRs [
17,
21], as well as to hybrid receptors containing InsR subunits with IGF-1R subunits [
22]. Both receptor types, however, use similar downstream signaling machinery [
23,
24,
25,
26]. Consequently, discerning actions of insulin in the brain through InsRs vs. IGF-1Rs is vital for understanding insulin’s effect on brain function and for specific targeting of appropriate receptor-mediated signaling components to combat brain dysfunction.
2. Insulin Promotes Striatal Dopamine Release through ChIs and nAChRs
Striatal dopamine release is governed by the activity of midbrain dopamine neurons, but can also be triggered from within the striatum by activation of nicotinic acetylcholine receptors (nAChRs) that are enriched on dopamine axons [
40,
41,
42,
43]. The source of acetylcholine (ACh) for this process are cholinergic interneurons (ChIs), which make up only a small percentage of the neuronal population of the striatum, yet exert a powerful influence on dopamine release and striatal function through extensive axonal processes that enable non-synaptic, as well as synaptic-like, transmission with dopamine axons [
43].
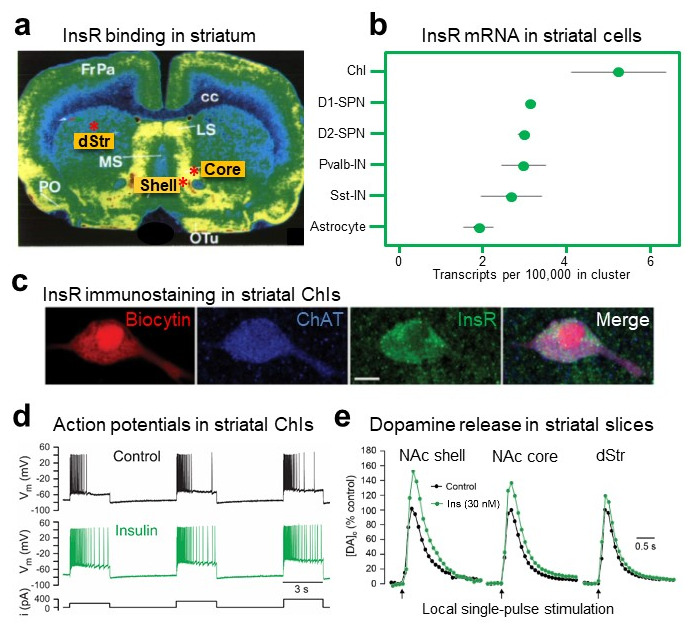
Early studies using autoradiography show abundant InsR binding sites throughout the striatum (
Figure 1a) [
15,
16,
17] but lack the resolution to discern which striatal cells express these InsRs. Analysis of RNA-seq data in DropViz (
http://dropviz.org, accessed on 16 September 2022) [
44], shows the highest levels of mRNA for InsRs in ChIs compared to other striatal cells including striatal projection neurons (SPNs), that are the most prevalent neuronal population in the striatum (
Figure 1b). Consistent with abundant mRNA in ChIs, immunohistochemical examination of InsRs in rat striatum indicates dense labeling for InsR proteins in ChIs, identified by co-immunostaining for the primary enzyme for ACh synthesis, choline acetyltransferase (ChAT) (
Figure 1c) [
19]. Thus, ChIs provide a significant target by which insulin can influence dopamine transmission.
Figure 1. InsR localization in striatum and insulin-dependent regulation of striatal dopamine release via ChIs, InsRs and nAChRs. (
a) Autoradiographic image of insulin receptor (InsR) binding sites throughout the rat striatum, including in the dorsal striatum (dStr), nucleus accumbens (NAc) core and shell subregions marked by red asterisks; modified with permission from [
16]. Abbreviations: cc, corpus callosum; FrPa, frontal parietal cortex; LS, lateral septum; MS, medial septum; PO, preoptic area; oTu, olfactory tubercle. (
b) Comparison of relative mRNA levels for InsRs in striatal cells shows high abundance in cholinergic interneurons (ChIs) versus dopamine D1 or D2 spiny projection neurons (SPNs), parvalbumin positive interneurons (Pvalb-IN), somatostatin positive interneurons (Sst-IN) or astrocytes; RNA-seq data was obtained using DropViz. (
c) Immunohistochemical staining of InsRs in a biocytin-filled, ChAT-positive ChI; scale = 10 μm; images from [
19]. (
d) Response of current-clamped rat ChI to depolarizing current pulses; insulin (30 nM) increases ChI excitability by attenuating action potential (AP) accommodation; from [
19]. (
e) Insulin (30 nM) enhances single-pulse evoked increases in extracellular dopamine concentration ([DA]
o) in dStr but has a greater effect in NAc core and NAc shell subregions of rat striatal slices; from [
19].
2.1. Insulin Increases the Excitability of Striatal ChIs
Electrophysiological recordings in
ex vivo striatal slices have demonstrated functional consequences of InsR activation in ChIs. The method used is whole-cell recording, in which a micropipette containing a solution resembling the cytoplasm forms a continuum with an individual cell, enabling the electrical activity of the cell to be monitored. In experiments using current-clamp recording in which the current across a cell membrane is controlled and membrane potential is monitored, injecting positive current to depolarize a recorded neuron causes an initial burst of action potentials (APs) that shows a progressive decrease in frequency with continued depolarization. This is referred to as AP accommodation [
45]. Addition of a physiological concentration of insulin (30 nM) to the artificial cerebrospinal fluid (aCSF) that superfuses the slice increases ChI excitability, seen as an increase in AP number during prolonged depolarization, i.e., attenuated AP accommodation (
Figure 1d) [
19]. This increase in excitability is InsR dependent, as the effect of insulin is prevented by an InsR inhibitor, whereas blocking IGF-1Rs has no effect [
19]. The mechanism by which insulin increases ChI excitability is unresolved, but is likely to involve one or more ion channels governing the membrane dynamics of these neurons [
45,
46].
2.2. Insulin Boosts Dopamine Release via InsRs, and Requires PI3K and nAChRs
The action of insulin on ChI excitability is sufficient to have a profound impact on axonal dopamine release in striatal slices as monitored using fast-scan cyclic voltammetry (FSCV). FSCV is an electrochemical method used to monitor the profile of evoked increases in extracellular dopamine concentration ([DA]
o) by detecting the current generated during the oxidation of dopamine at the surface of a carbon fiber microelectrode when a triangular voltage waveform is applied. The voltage waveform is typically applied every 100 ms and the carbon fiber microelectrode is typically 5 to 10 μm in diameter thereby providing a dopamine release profile with high temporal (subsecond) and high spatial (few micrometer) resolution [
47,
48]. Previous studies using FSCV show that up to 70% of dopamine release evoked by a single electrical pulse in striatal slices can be attributed to ChI-induced ACh release and consequent activation of nAChRs on dopamine axons [
49,
50,
51,
52]. Strikingly, application of low nM insulin via the aCSF amplifies single pulse-evoked [DA]
o further, with a greater enhancement in the reward-related nucleus accumbens (NAc) core and shell regions of the ventral striatum than in the motor-related dorsal striatum (dStr) (
Figure 1e). This regional heterogeneity in the response to insulin mirrors the regional density of InsRs in the striatum (
Figure 1a) [
15,
16,
17]. Showing that the influence of insulin on dopamine release involves dynamic regulation of the exocytotic process, tissue dopamine content of striatal slices is unaltered when exposed to 30 nM insulin, eliminating potential contributions from changes in dopamine synthesis or storage [
19].
Like the changes seen in ChI excitability, amplification of evoked [DA]
o by insulin is mediated by InsRs rather than IGF-1Rs; insulin-induced increases in dopamine release are prevented by blocking InsRs, but not by blocking IGF-1Rs [
19]. Interestingly, a higher concentration of insulin, 100 nM, does not produce a significant enhancement of evoked dopamine release which could reflect competing activation of IGF-1Rs, as seen in SPNs, although this has not been investigated.
InsRs are members of the receptor tyrosine kinase superfamily which activate two major signaling pathways: the phosphatidylinositol 3-kinase (PI3K) pathway and the mitogen-activated protein kinase (MAPK) pathway [
53]. Insulin-induced changes in evoked [DA]
o are prevented in the presence of a PI3K inhibitor in all striatal subregions, revealing the involvement of known downstream signaling mechanisms for insulin activation of InsRs in this process [
19]. Demonstrating a pivotal ACh-DA interaction, the effect of insulin on evoked [DA]
o is prevented by nAChR antagonists [
19]. Insulin amplifies evoked striatal [DA]
o in slices from mice, as well as from rats; however, the effect of insulin is absent throughout the striatum of mice with genetic deletion of
ChAT (
ChAT KO) [
19]. Together these observations provide a model by which insulin activation of InsRs on ChIs induces enhancement of ChI activity which then boosts ACh-mediated dopamine release via nAChRs on dopamine axons (
Figure 2).
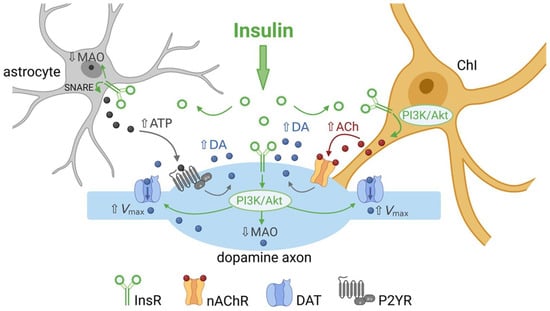
Figure 2. Summary of the multiple ways by which insulin can regulate striatal dopamine (DA) transmission. Activation of insulin receptors (InsRs) on cholinergic interneurons (ChIs) increases ChI excitability, which increases acetylcholine (ACh) release and activation of nicotinic ACh receptors (nAChRs) on dopamine axons to boost dopamine release and increase extracellular DA concentration ([DA]o). Activation of InsRs on dopamine axons increases dopamine transporter (DAT) surface expression via the PI3K/Akt pathway to increase the maximum velocity (Vmax) of dopamine uptake, thereby lowering [DA]o. Activation of InsRs on astrocytes increases exocytotic ATP release, which activates purinergic P2Y receptors (P2YR) on dopamine axons and enhances dopamine release. Insulin also decreases monoamine oxidase (MAO) activity in dopamine axons and glial cells, thereby decreasing dopamine metabolism and prolonging the time course of dopamine actions. Illustration created with BioRender.com.
3. Insulin Enhances Striatal Dopamine Uptake via DAT on Dopamine Axons
The dopamine transporter (DAT) is expressed exclusively in dopamine neurons in both the midbrain cell body region and forebrain dopamine axons in the striatum [
54,
55,
56] and is crucial for curtailing the amplitude and duration of dopamine transients, as well as maintaining homeostatic levels of [DA]
o through transporter-mediated uptake [
57,
58]. Thus, [DA]
o at a given location and time reflects a balance between the opposing processes of release and uptake [
51,
52]. In addition, DAT-dependent dopamine uptake provides a major source of dopamine for vesicular uptake and storage for subsequent release [
59].
In addition to the localization of InsRs in striatal cells, immunohistochemical analysis of striatal InsRs show expression on dopamine axons, identified by the presence of tyrosine hydroxylase, the primary enzyme required for dopamine synthesis [
19]. Downstream effects of InsR activation include the regulation of dopamine uptake. Insulin increases expression of mRNA for DAT in midbrain dopamine neurons [
60], which could theoretically increase total DAT protein levels. However, the most profound effect that insulin has on the DAT is its ability to redistribute DAT localization within a dopamine neuron. Although an integral part of the plasma membrane, DAT surface expression is dynamically regulated by endocytic trafficking mediated by a variety of intracellular signaling pathways, including PKC, which promotes DAT internalization, and the PI3K-Akt signaling pathway which has the opposite effect [
61,
62,
63]. Given that both InsRs and IGF-1Rs belong to the receptor tyrosine kinase superfamily that couple to PI3K via phosphorylation of InsR substrates [
23,
24,
25,
26], insulin can influence DAT surface expression and function via these pathways. In striatum, it has been shown that in addition to enhancing dopamine release, insulin promotes DAT trafficking to the plasma membrane through PI3K-Akt signaling, thereby increasing overall DAT activity and dopamine uptake [
61,
62,
64,
65,
66]. Dopamine uptake in the striatum is governed by Michaelis–Menten kinetics [
67] and has been assessed using several different methods and experimental preparations. Although the values for the uptake kinetic parameters obtained are not directly comparable across studies [
68], the general consensus is that insulin increases the maximal uptake velocity,
Vmax, for dopamine with little or no effect on
Km, which is inversely related to the affinity of the DAT for dopamine [
61,
64,
65,
66,
69]. Moreover, although dopamine uptake is less efficient in the ventral striatum than in the dorsal striatum [
70,
71,
72], enhanced dopamine uptake by insulin is seen throughout the striatal complex and in both rats and mice [
64,
65,
66,
69].
Confirming activation of the PI3K-Akt pathway, insulin-induced increases in striatal DAT activity are prevented by PI3K inhibitors [
61,
64,
66,
69]. In addition, it has been shown that inhibiting PI3K alone can decrease
Vmax for dopamine uptake into synaptosomes (isolated membrane bound axonal segments containing vesicles and mitochondria) implying that this pathway tonically regulates DAT activity [
61]. However, in studies using striatal slices, PI3K inhibition prevents insulin-mediated increases in dopamine uptake without altering tonic regulation of DAT function [
66]. Notably, although PI3K can be activated by insulin acting at either InsRs or IGF-1Rs [
53], blocking InsRs occludes the effect of 30 nM insulin on dopamine uptake, whereas blocking IGF-1Rs does not [
66]. Thus, insulin-induced enhancement of dopamine uptake in the striatum is mediated by InsRs and the PI3K pathway in dopamine axons (
Figure 2).
The enhancing effect of insulin on dopamine uptake would be expected to decrease evoked [DA]
o. However, as described in
Section 2, studies using FSCV detect elevated increases in [DA]
o in the presence of insulin [
19,
69], which reflects the dominant effect of insulin on increasing ACh-induced dopamine release. When striatal ACh is absent, as in
ChAT-KO mice [
50], insulin’s action on dopamine uptake prevails, revealing a net decrease in evoked [DA]
o with insulin application [
66]. Given that DAT is a mediator of the short-term plasticity of dopamine release [
73], insulin’s actions on DAT expression and dopamine uptake will have implications for overall dopamine transmission, independent of the influence of ChIs on dopamine release.
4. Insulin Inhibits Dopamine Metabolism
In addition to enhancing dopamine release through nAChRs and increasing dopamine uptake through the DAT, there is evidence that insulin can alter dopamine metabolism [
31,
74,
75]. Key metabolizing enzymes for dopamine are monoamine oxidase enzymes (MAOs), which may be located in dopamine axons or in astrocytes (see
Figure 2). In rodents, MAO mainly catalyzes the formation of the dopamine metabolite dihydroxyphenylacetic acid (DOPAC). Samples of striatal extracellular fluid taken using microdialysis in freely moving rats show a profound and long-lasting decrease in DOPAC levels following a peripheral insulin injection [
74]. Moreover, mice with brain-wide deletion of InsRs have increased MAO expression in the striatum [
75]. Consequently, total area under the curve for single-pulse evoked [DA]
o in the dStr and NAc is lower in striatal slices from mice lacking brain InsRs than from controls. Although peak amplitude was the same between genotypes, the overall duration of the release response was shortened [
75]. This suggests another route by which endogenous insulin could enhance [DA]
o, through inhibition of dopamine metabolism via MAO suppression (
Figure 2).
5. Insulin Bidirectionally Alters the Excitatory Regulation of Spiny Projection Neurons (SPNs)
In addition to its effect on ChIs and dopamine axons, insulin also acts at other striatal neurons including SPNs, thereby influencing overall striatal output. These projection neurons represent over 95% of the total neuronal population of the striatum, and can be subdivided into two classes based on expression of dopamine D1 vs. dopamine D2 receptor subtypes, as well as differential expression of endogenous opioid peptides, and different projection targets [
76,
77]. However, levels of InsR-mRNA appear to be similar in the two classes of SPNs (
Figure 1b). Whole-cell voltage-clamp recordings of electrically evoked excitatory post-synaptic currents (eEPSCs) from individual SPNs in striatal slices show that 30 nM insulin increases the amplitude of eEPSCs via activation of InsRs on both D1- and D2-SPNs within the NAc core. By contrast, insulin concentrations ≥100 nM have the opposite effect of decreasing eEPSC amplitude through activation of IGF-1Rs [
78]. Moreover, IGF-1Rs appear to provide tonic inhibition of SPNs because blocking IGF-1Rs with PPP alone increases eEPSC amplitude and also enhances the effect of 30 nM insulin. Analysis of the effect of 30 nM insulin on spontaneous miniature EPSCs (mEPSCs) show an increase in the frequency of these events, but not the amplitude, as well as a decrease in paired-pulse-ratio; all are consistent with enhanced excitatory transmission mediated by a boosting of presynaptic glutamate release. Again, a higher concentration of insulin, 100 nM, has the opposite effect through activation of IGF-1Rs, suggesting that IGF-1R-mediated reductions in excitatory transmission are also due to effects on presynaptic glutamate release [
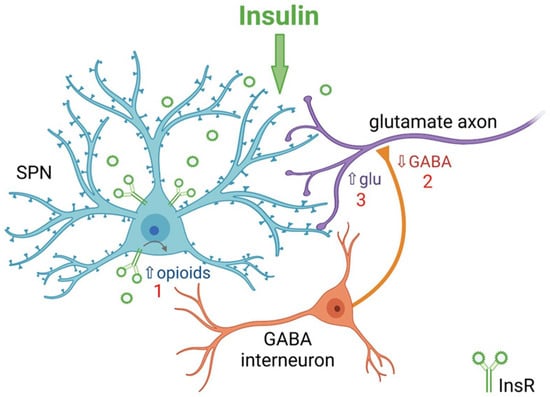
78]. Thus, insulin causes bidirectional effects on SPN excitability through activation of InsRs and IGF-1Rs. The mechanisms by which insulin modulates SPN activity appears to involve a feedback microcircuit in which insulin enhances SPN excitability, through activation of InsRs on SPNs and subsequent release of endogenous opioids, which then inhibit local GABAergic interneurons leading to disinhibition of presynaptic glutamate release that ultimately influences striatal output (
Figure 3) [
78].
Figure 3. Summary of mechanism by which insulin increases excitatory regulation of striatal projection neurons (SPNs) in nucleus accumbens (NAc) core. 1. Activation of insulin receptors (InsRs) on GABA/opioid containing SPNs in rat NAc core increases the release of endogenous opioids. 2. Opioid activation of GABAergic interneurons decreases GABA release onto glutamatergic inputs from cortex and thalamus. 3. Decreased GABA release from GABAergic interneurons disinhibits glutamatergic inputs resulting in increased presynaptic glutamate (Glu) release onto dendritic spines of SPNs. Higher concentrations of insulin act at insulin-like growth factor-1 receptors (IGF-1Rs) to oppose this process (not shown). Illustration created with BioRender.com.
6. Insulin Actions on Striatal Glial Cells
Astrocytes, like other glial cells in the brain (microglia and oligodendrocytes), play an important role in maintaining homeostasis of the brain microenvironment during all stages of life. This is achieved through maintaining the BBB, supplying trophic support to neurons, regulating local ionic concentrations, mediating synapse formation and function, pruning synapses during development and signaling across brain regions to modulate neuronal activity [
79,
80,
81]. Moreover, astrocytes are instrumental in controlling levels of a variety of neurotransmitters/neuromodulators, including the key transmitters glutamate and GABA, as well as releasing these transmitters and other gliotransmitters [
82,
83,
84,
85].
Brain insulin has been shown to play an important role in regulating systemic glucose metabolism through astrocytic InsRs in the hypothalamus [
85,
86,
87,
88]. Moreover, insulin can act on astrocytes and other glial cells to regulate local cellular metabolism [
89]. DropViz data show that striatal astrocytes express InsR mRNA (
Figure 1b). Mice with astrocyte-specific InsR deletion exhibit a decrease in the peak amplitude of evoked [DA]
o in both dStr and NAc with no change in response duration, or in MAO levels [
90]. The mechanism by which InsRs in astrocytes regulate dopamine release has been proposed to be through targeting tyrosine phosphorylation of SNARE proteins that enable exocytotic ATP release, which in turn enhances axonal dopamine release via G-protein coupled P2Y purinergic receptors (
Figure 2) [
35,
90]. These findings add to the range of mechanisms through which insulin can alter striatal dopamine signaling.
7. Conclusions
It is clear that insulin’s actions in the striatum are multifaceted in terms of the striatal elements involved and which insulin-sensitive receptors mediate the effects. Insulin can boost dopamine release indirectly through actions at InsRs on ChIs and astrocytes, with slower regulation by inhibiting dopamine metabolism by MAO. In addition, insulin actions on SPNs that engage local striatal microcircuits can have a direct influence on striatal and basal ganglia output. Information about the behavioral impact of striatal insulin signaling is limited. However, the few studies available have provided valuable evidence indicating an important role for striatal insulin beyond its role as a satiation/satiety signal.