1. Introduction
In its healthy state, the liver functions as an immune sentinel, sampling the blood that enters it via the hepatic portal vein, before it reaches the spleen or lymph nodes. This blood is rich in nutrients, as well as any pathogen-derived molecules that are in circulation, such as lipopolysaccharide (LPS). Since the liver filters all of the blood, it is in a prime position to detect these molecules and sound the alarm to the immune system. It is, therefore, unsurprising that damage to the liver leads to a robust immune response, by both the innate and adaptive arms of the immune system (
Figure 1). NAFLD leads to lipotoxicity within the liver, which, in turn, causes hepatocytes to become stressed or die, and this process generates inflammatory factors called damage-associated molecular patterns (DAMPs) [
130], which are able to trigger activation of the immune system [
131]. Whilst the precise mechanisms leading to the progression from NAFLD to NASH remain unclear, there is evidence to suggest that these are, in part, immunological in nature.
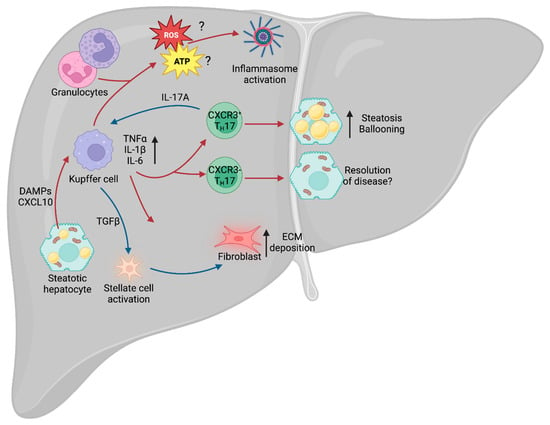
Figure 1. Liver immunity. Schematic of the broad immune changes occurring in the liver during NAFLD. Hepatic steatosis causes the release of DAMPs and CXCL10, leading to activation of the liver-resident macrophages (Kupffer cells). Kupffer cells, in turn, recruit the adaptive arm of the immune system, including TH17 cells, which can lead to exacerbation of NAFLD. Additionally, IL-17A stimulates secretion of TGFβ by Kupffer cells, which drives stellate cell activation and release of ECM from fibroblasts. Kupffer cell-derived ROS and ATP also contribute to NAFLD- and NASH-associated inflammation. NAFLD, non-alcoholic fatty liver disease; DAMPs, damage-associated molecular patterns; TH17, T helper 17 cell; IL-17A, TH17-derived interleukin 17A; ECM, extracellular matrix; ROS, reactive oxygen species; NASH, non-alcoholic steatohepatitis.
2. Immunity and NAFLD/NASH
Approximately 30% of patients with NAFLD develop an inflammatory phenotype and progress to NASH, with subsequent tissue injury and the development of hepatic fibrosis. However, this key step between the relatively benign NAFLD phenotype and inflammatory NASH remains subject to extensive debate. Chronic metabolic inflammation (metaflammation) is initially promoted during metabolic diseases such as obesity and type 2 diabetes (T2D), and there is evidence to suggest that this fuels the NAFLD–NASH transition. In particular, the adipose tissue has been identified as a major source of inflammatory cytokines, including pro-inflammatory tumor necrosis factor (TNF), interleukin-1β (IL-1β) and IL-6 [
132]. In mouse models of hepatic steatosis, systemic deletion of
Tnf and chemical inhibition of TNF-Receptor-1 reduces the prevalence of steatosis and hepatocellular injury [
133,
134]. At the hepatic level, lipotoxicity within hepatocytes drives the release of CXCL10 (C-X-C motif ligand 10) [
135], which is a chemoattractant, or CXCR3 (C-X-C motif chemokine receptor 3)-expressing Kupffer cells [
136], the tissue-resident macrophages of the liver. Upon localization to the site of injury and stimulation of toll-like receptor 4 (TLR4), Kupffer cells also release TNF, IL-1β and IL-6 [
137]. TNF-alpha (TNFα) is a major contributor to the inflammatory response, regulating various aspects of sickness behavior during infection, including fever and cachexia. In the context of NAFLD, TNFα was shown to drive an increase in the expression of the genes
Acaca (acetyl-CoA carboxylase alpha) and
Scd1 (stearoyl-CoA desaturase 1) [
133].
Acaca encodes a rate-limiting enzyme for fatty acid synthesis [
138], whilst
Scd1 encodes a lipogenic enzyme that catalyzes the synthesis of monounsaturated fatty acids [
139]. Furthermore, inhibition of the TNFα receptor, TNFR1, improves liver steatosis and insulin resistance [
134]. Given that TNFα is a major driver of cachexia during infection, it is paradoxical that it also drives increased expression of these two lipid storage enzymes in NAFLD, suggesting that the role of this cytokine is tissue- and context-dependent.
IL-6 is an important driver of adaptive immune recruitment, selectively controlling T cell recruitment by mediating chemokine secretion [
140]. Further to recruitment of T cells, IL-6 also drives polarization of CD4
+ T cells, inhibiting T helper 1 (T
H1) and promoting T
H2 and T
H17 differentiation [
141,
142]. In both NAFLD and NASH, patients exhibit increased circulating IFNγ-producing T
H1 cells, and patients with NASH could be stratified from those with NAFLD by the increase in circulating T
H17 cells [
143]. T
H17 cells are CD4
+ cells that express the transcription factor RORγt (retinoid orphan receptor gamma t) and RORα, and are characterized by secretion of IL-17A and IL-17F. It was recently demonstrated that IL-17A and IL-17F are drivers of adipocyte lipid usage in adipocytes during infection and, moreover, promote infection-induced cachexia [
144], suggesting that it may drive lipid usage in other cell types, including hepatocytes. As with TNFα, this appears paradoxical, as NAFLD is associated with increased hepatic lipid storage. However, increased expression of TNFα and IL-17A may represent a mechanism by which the liver attempts to mobilize and dispose of excess lipids to restore homeostatic function.
T
H17 cells are known to expand in the liver of obese humans and mice [
145], and multiple rodent models of NAFLD show increased IL-17A signaling through the IL-17A receptor (IL-17RA) [
146,
147]. The development of NAFLD leads to increased infiltration of non-conventional CXCR3
+ T
H17 cells, which can co-express IFNγ. Adoptive transfer of CXCR3
+ T
H17 cells into mice with experimental NAFLD increased hepatic damage compared with those given CXCR3
− T
H17 cells [
148]. Furthermore, the livers of mice given CXCR3
+ T
H17 cells displayed increased triglyceride accumulation and hepatocyte ballooning. Moreover, the presence of CXCR3
+ T
H17 cells correlated with increased disease severity in humans, suggesting that this is an evolutionarily conserved aspect of NAFLD and NASH. However, the mechanisms by which these CXCR3
+ T
H17 cells exacerbate disease remain unclear. Typically, T
H17 cell-secreted IL-17A recruits and activates neutrophils [
149], a cell type abundant in the liver of NASH patients, but it remains to be seen whether CXCR3
+ T
H17 cells exert their effects through neutrophil activation or an as yet unknown mechanism. Further to its role in recruitment of neutrophils, IL-17A signals through IL-17RA on Kupffer cells to promote production of TGF-β1 (transforming growth factor β1) [
150], which, in turn, promotes hepatic stellate cell (HSC) activation and extracellular matrix secretion, contributing to NAFLD progression [
151].
3. Immunometabolism in NAFLD/NASH
An emerging area of interest in NAFLD and NASH is immunometabolism. All cells rely on nutrients to function and immune cells are no exception. Indeed, during infection and injury, the immune system requires significant amounts of energy to fuel itself, with different cell types relying on specific nutrients for optimal function. Activation of Kupffer cells leads to enhanced glucose utilization [
152] and a rapid increase in aerobic glycolysis (the “Warburg Effect”), whereby cells preferentially rely on glycolysis despite the presence of oxygen [
153]. Although glycolysis is less energetically favorable than oxidative phosphorylation (OXPHOS) [
154], it carries a number of advantages for immune cells. Early branching of the glycolytic pathway generates precursors for the pentose phosphate pathway (PPP) and de novo nucleotide synthesis, which are required for the function of multiple immune cell types [
155]. For example, in macrophages, such as Kupffer cells, the PPP is required to sustain superoxide anion production [
156], a key component for the phagocytic oxidative burst [
157], which is a tool for destroying pathogens. In the context of NAFLD, a wide variety of cells increase superoxide production, including the aforementioned Kupffer cells, as well as neutrophils and other granulocytes [
158], which may contribute to NAFLD-associated oxidative stress. Furthermore, NAFLD is associated with increased aerobic glycolysis [
159], leading to increased lactate production and stabilization of HIF-1α (hypoxia-inducible factor 1α). This HIF-1α stabilization, in turn, promotes usage of the glycolytic pathway and promotes liver fibrosis in murine models of NAFLD [
160], potentially signaling a key step in the transition from NASH to cirrhosis.
A further aspect of the immune system associated with NAFLD and progression to NASH is the NLRP3 (NOD-, LRR- and pyrin domain-containing protein 3) inflammasome. The NLRP3 inflammasome is an intracellular structure that senses microbial compounds and environmental stress. Its assembly leads to production of IL-1β and IL-18, as well as caspase-1-dependent apoptosis [
161]. In mouse models of NASH, administration of a selective NLRP3 inhibitor suppressed caspase-1 and IL-1β accumulation, and limited the development of fibrosis [
162], suggesting that the inflammasome plays a key role in mediating the progression of NAFLD. One of the major drivers of the inflammasome is ROS (reactive oxygen species) production, generated by OXPHOS. The mitochondrial electron transport chain (ETC) is an essential structure for ATP generation and is composed of multiple complexes (complexes I, II, III, IV and ATPase). Complex II, also known as succinate dehydrogenase, links the ETC with the TCA cycle, and chemical inhibition of this subunit prevents NLRP3 inflammasome activation [
163]. Whilst there is evidence to suggest that ROS production leads to activation of the NLRP3 inflammasome [
164], recent work suggests that it is ATP generation, and not ROS, that is required for inflammasome activation [
163]. Since NAFLD and NASH are prevalent in patients with concurrent obesity and metabolic dysfunction, increased metabolic cycling through glycolysis and OXPHOS may exacerbate inflammasome activation and oxidative damage.
Understanding the immune response mechanism in NAFLD is crucial for developing a complete picture of sexual dimorphism in liver disease. This gap in our knowledge needs addressing as it has critical implications for the development of new therapeutics.
This entry is adapted from the peer-reviewed paper 10.3390/cells12121604