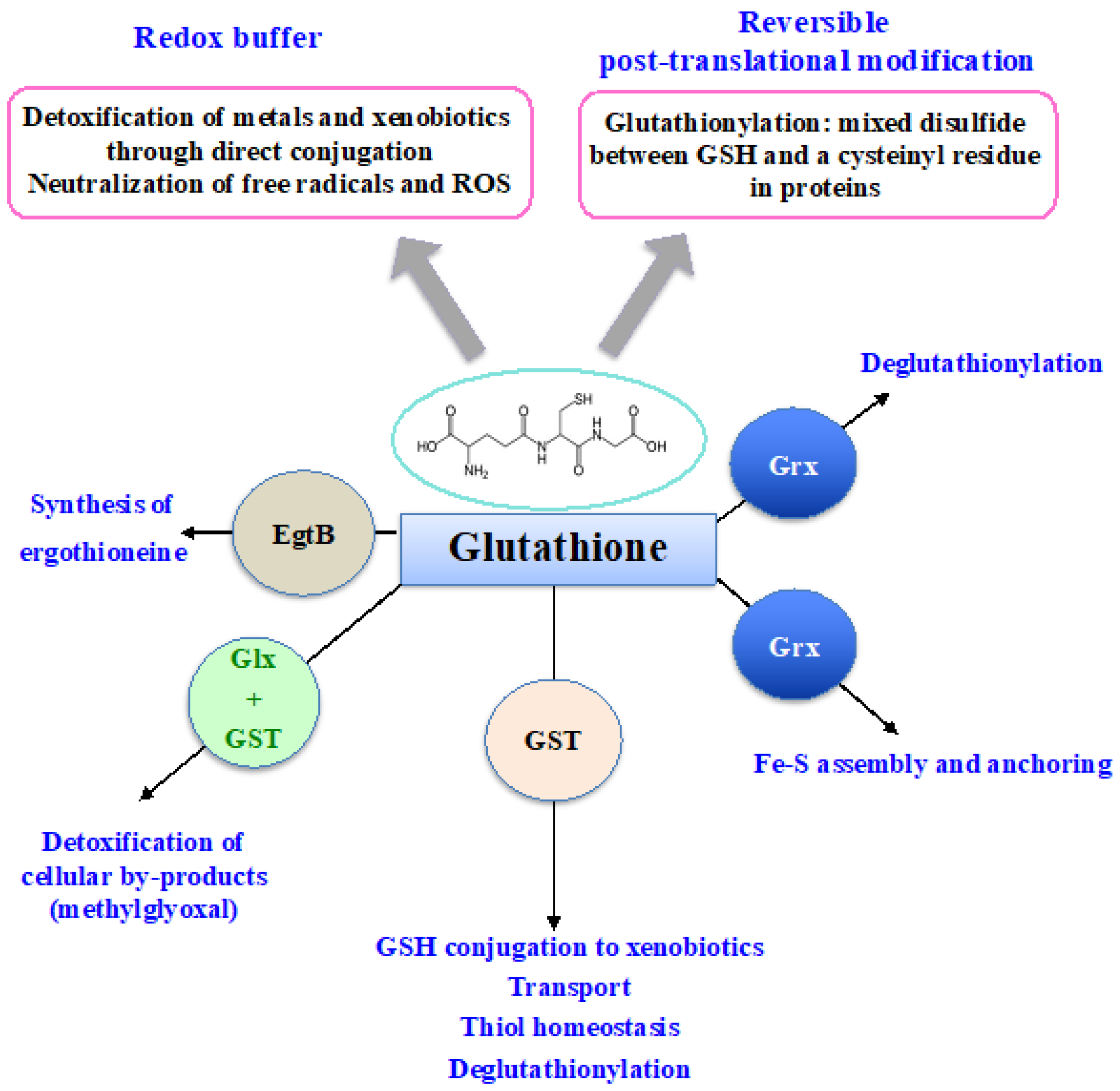
From bacteria to plants and humans, the glutathione system plays a pleiotropic role in cell defense against metabolic, oxidative and metal stresses. Glutathione (GSH), the γ-L-glutamyl-L-cysteinyl-glycine nucleophile tri-peptide, is the central player of this system that acts in redox homeostasis, detoxification and iron metabolism in most living organisms. GSH directly scavenges diverse reactive oxygen species (ROS), such as singlet oxygen, superoxide anion, hydrogen peroxide, hydroxyl radical, nitric oxide and carbon radicals. It also serves as a cofactor for various enzymes, such as glutaredoxins (Grxs), glutathione peroxidases (Gpxs), glutathione reductase (GR) and glutathione-S-transferases (GSTs), which play crucial roles in cell detoxication.
- cyanobacteria
- human
- plants
- glutathione
- glutaredoxins
- methylglyoxal
- ophthalmate
- norophthalmate
- glutathione-S-transferases
- iron-sulfur cluster
- Oxidative stress
- ergothioneine
1. Introduction
2. Biological Importance and Biotechnological Interests of Cyanobacteria
3. Synthesis and Importance of Glutathione in Living Organisms
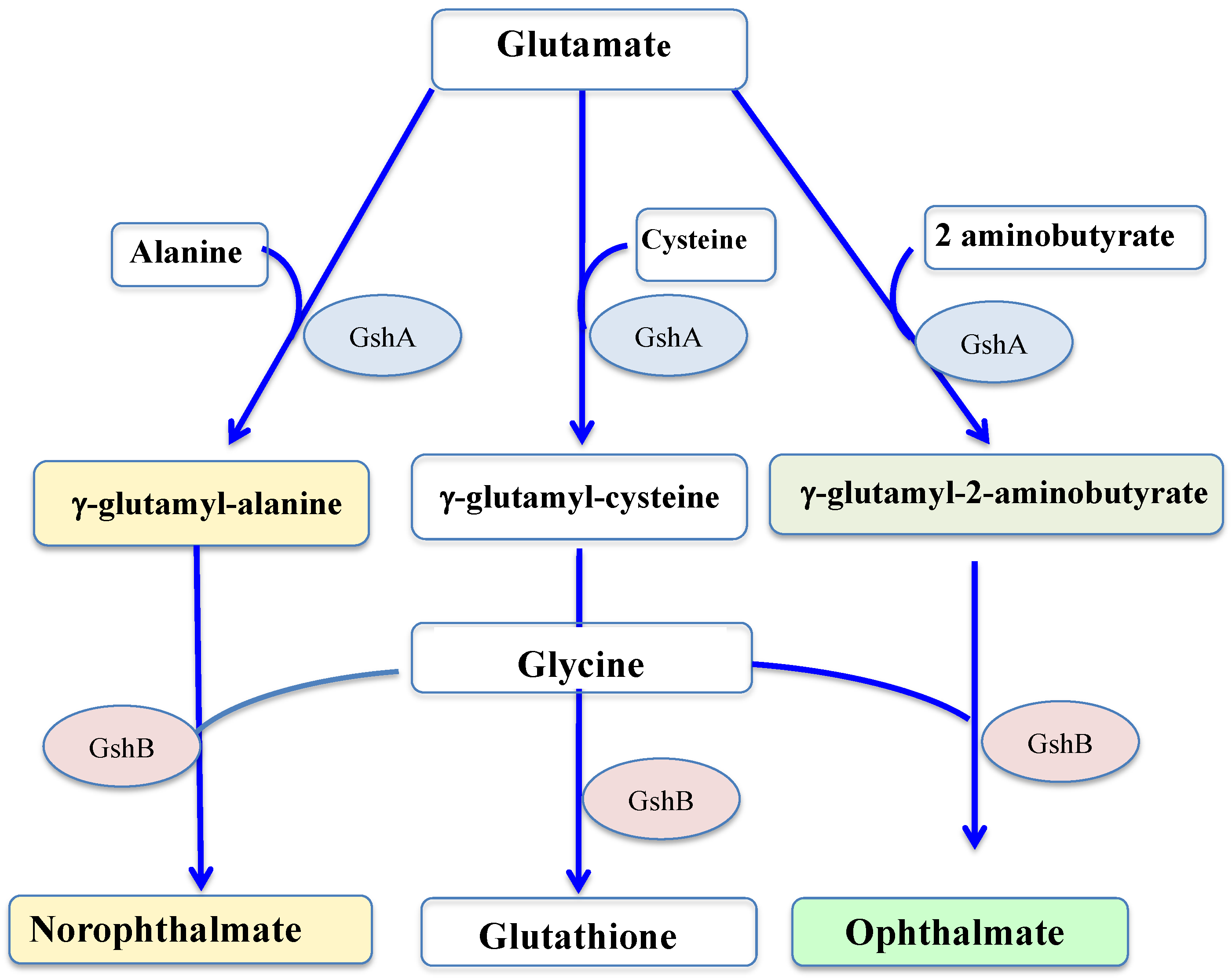
4. Glutathione Degradation
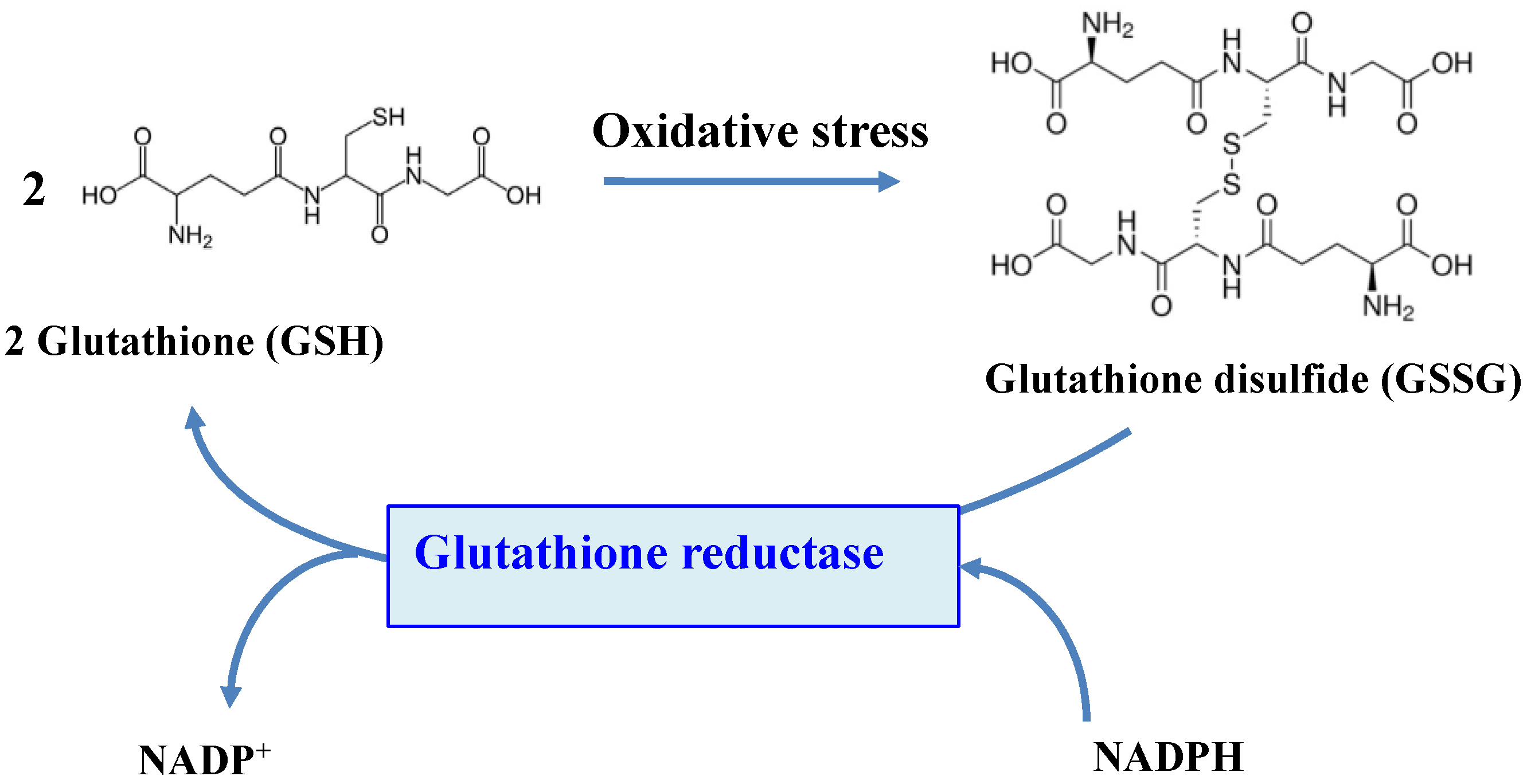
5. Glutathione Reductase
6. Importance of the Evolutionary-Conserved Glutathione-S-Transferase Enzymes
7. Glutathione Acts in the Detoxification of Methylglyoxal, a By-Product of Cell Metabolism from Cyanobacteria to Humans
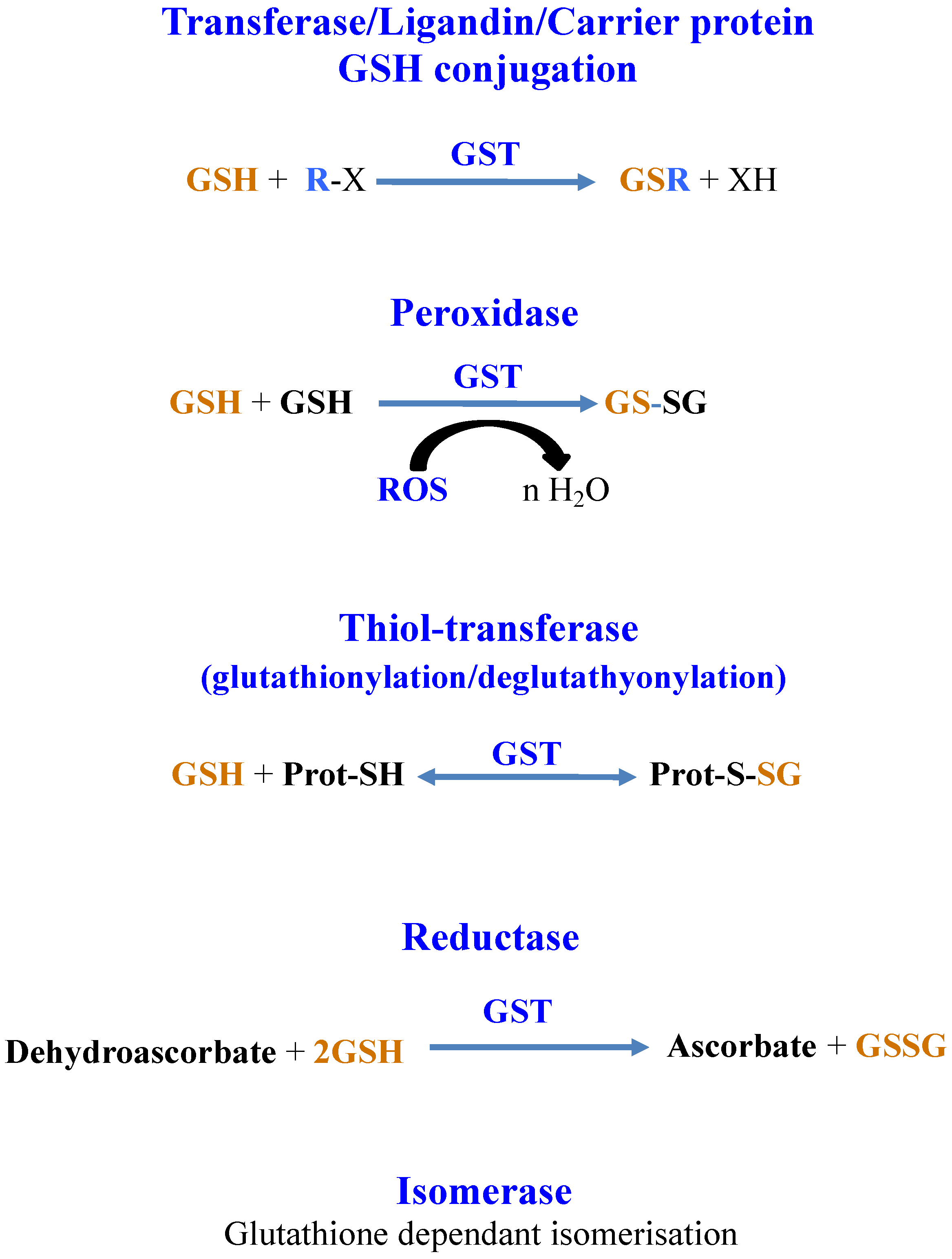
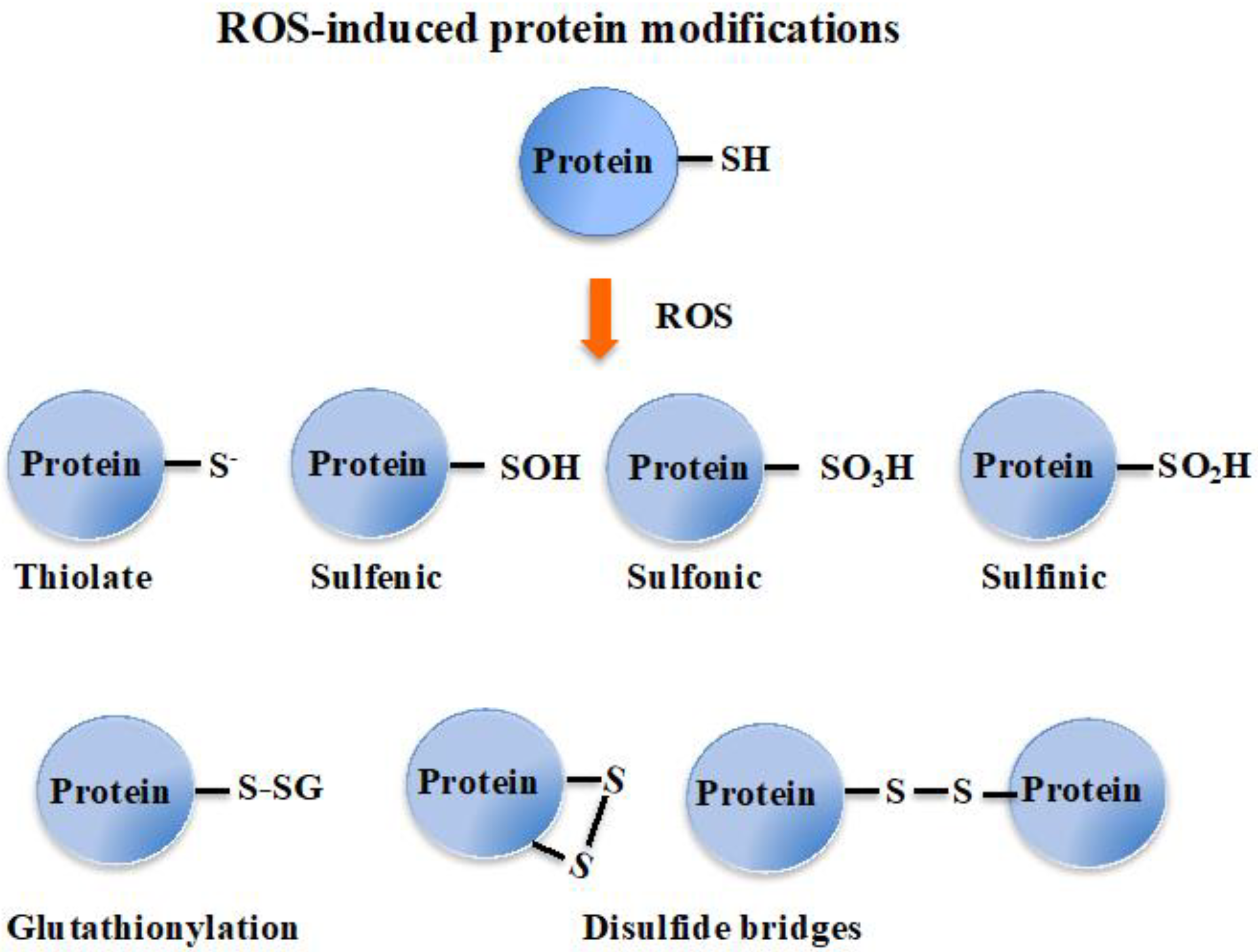
8. Glutathione Maintains the Redox Homeostasis of Protein Thiols via Glutathionylation/Deglutathionylation Catalyzed by Glutathione-S-Transferases and Some Glutaredoxins
9. Glutathione, Glutaredoxins and the Biogenesis of the Iron-Sulfur Cluster of Proteins
This entry is adapted from the peer-reviewed paper 10.3390/antiox12061199
References
- Vaish, S.; Gupta, D.; Mehrotra, R.; Mehrotra, S.; Basantani, M.K. Glutathione S-transferase: A versatile protein family. 3 Biotech 2020, 10, 321.
- Imlay, J.A. The molecular mechanisms and physiological consequences of oxidative stress: Lessons from a model bacterium. Nat. Rev. Microbiol. 2013, 11, 443–454.
- Deponte, M. Glutathione catalysis and the reaction mechanisms of glutathione-dependent enzymes. Biochim. Biophys. Acta Gen. Subj. 2013, 1830, 3217–3266.
- Cameron, J.C.; Pakrasi, H.B. Essential role of glutathione in acclimation to environmental and redox perturbations in the cyanobacterium Synechocystis sp. PCC 6803. Plant Physiol. 2010, 154, 1672–1685.
- Fahey, R.C. Glutathione analogs in prokaryotes. Biochim. Biophys. Acta Gen. Subj. 2013, 1830, 3182–3198.
- Musgrave, W.B.; Hankuil, Y.; Kline, D.; Cameron, J.C.; Wignes, J.; Dey, S.; Pakrasi, H.B.; Jez, J.M. Probing the origins of glutathione biosynthesis through biochemical analysis of glutamate-cysteine ligase and glutathione synthetase from a model photosynthetic prokaryote. Biochem. J. 2013, 450, 63–72.
- Kammerscheit, X.; Chauvat, F.; Cassier-Chauvat, C. First in vivo evidence that glutathione-S-transferase operates in photo-oxidative stress in cyanobacteria. Front. Microbiol. 2019, 10, 1899.
- Rai, R.; Singh, S.; Rai, K.K.; Ral, A.; Sriwastaw, S.; Rai, L.C. Regulation of antioxidant defense and glyoxalase systems in cyanobacteria. Plant Physiol. Biochem. 2021, 168, 353–372.
- Bachhawat, A.K.; Yadav, S. The glutathione cycle: Glutathione metabolism beyond the γ-glutamyl cycle. IUBMB Life 2018, 70, 585–592.
- Noctor, G.; Reichheld, J.-P.; Foyer, C.H. ROS-related redox regulation and signaling in plants. Semin. Cell Dev. Biol. 2018, 80, 3–12.
- Dorion, S.; Ouellet, J.C.; Rivoal, J. Glutathione Metabolism in Plants under Stress: Beyond Reactive Oxygen Species Detoxification. Metabolites 2021, 11, 641.
- He, Y.-Y.; Hader, D.-P. Reactive oxygen species and UV-B: Effect on cyanobacteria. Photochem. Photobiol. Sci. 2002, 1, 729–736.
- Nawkar, G.N.; Maiban, P.; Park, J.H.; Sahi, V.P.; Lee, S.Y.; Kang, C.H. UV-Induced cell death in plants. Int. J. Mol. Sci. 2013, 14, 1608–1628.
- Sing, R.R.; Reindi, K.M. Glutathione S-Transferases in Cancer. Antioxidants 2021, 10, 701.
- Trnka, D.; Hossain, M.F.; Magdalena, J.L.; Gellert, M.; Lillig, C.H. Role of GSH and Iron-Sulfur Glutaredoxins in Iron Metabolism-Review. Molecules 2020, 27, 3860.
- Hristov, B.D. The Role of Glutathione Metabolism in Chronic Illness Development and Its Potential Use as a Novel Therapeutic Target. Cureus 2022, 14, e29696.
- Belenguer-Varea, A.; Tarazona-Santabalbina, F.J.; Avellana-Zaragoza, J.A.; Martinez-Reig, M.; Mas-Bargues, C.; Ingles, M. Oxidative stress and exceptional human longevity: Systematic review. Free Radic. Biol. Med. 2020, 149, 51–63.
- Deponte, M. The Incomplete Glutathione Puzzle: Just Guessing at Numbers and Figures? Antioxid. Redox Signal 2017, 27, 1130–1161.
- Forman, H.J.; Maiorino, M.; Ursini, F. Signaling Functions of Reactive Oxygen Species. Biochemistry 2010, 49, 835–842.
- Carmel-Harnel, O.; Storz, G. Roles of the Glutathione- and Thioredoxin-Dependent Reduction Systems in the Escherichia Coli and Saccharomyces cerevisiae Responses to Oxidative Stress. Annu. Rev. Microbiol. 2000, 54, 439–451.
- Masip, L.; Veeravalli, K.; Georgiou, G. The many faces of glutathione in bacteria. Antioxid. Redox Signal. 2006, 8, 753–762.
- Mailloux, R.J. Protein S-glutathionylation reactions as a global inhibitor of cell metabolism for the desensitization of hydrogen peroxide signals. Redox Biol. 2020, 32, 101472.
- Jena, A.B.; Samal, R.R.; Bhol, N.K.; Duttaroy, A.K. Cellular Red-Ox system in health and disease: The latest update. Biomed. Pharmacother. 2023, 162, 114606.
- Mallén-Ponce, M.J.; Huertas, M.J.; Florencio, F.J. Exploring the Diversity of the Thioredoxin Systems in Cyanobacteria. Antioxidants 2022, 11, 654.
- Kalinina, E.; Novichkova, M. Glutathione in Protein Redox Modulation through S-Glutathionylation and S-Nitrosylation. Molecules 2021, 26, 435.
- Ogata, F.T.; Branco, V.; Vaie, F.F.; Coppo, L. Glutaredoxin: Discovery, redox defense and much more. Redox Biol. 2021, 43, 101975.
- Nianiou-Obeidat, I.; Madesis, P.; Kissoudis, C.; Voulgari, G.; Chronopoulou, E.; Tsaftaris, A.; Labrou, N.E. Plant glutathione transferase-mediated stress tolerance: Functions and biotechnological applications. Plant Cell Rep. 2017, 36, 791–805.
- Gallé, Á.; Czékus, Z.; Bela, K.; Horváth, E.; Ördög, A.; Csiszár, J.; Poór, P. Plant glutathione transferases and light. Front. Plant Sci. 2019, 9, 1944.
- Musaogullari, A.; Chai, Y.C. Redox regulation by protein s-glutathionylation: From molecular mechanisms to implications in health and disease. Int. J. Mol. Sci. 2020, 21, 9113.
- Murata, K.; Kimura, A. Overproduction of glutathione and its derivatives by genetically engineered microbial cells. Biotechnol. Adv. 1990, 8, 59–96.
- Sanchez-Baracaldo, P.; Bianchini, G.; Wilson, J.D.; Knoll, A.H. Cyanobacteria and biogeochemical cycles through Earth history. Trends Microbiol. 2022, 30, 143–157.
- Berman-Frank, I.; Lundgren, P.; Falkowski, P. Nitrogen fixation and photosynthetic oxygen evolution in cyanobacteria. Res. Microbiol. 2003, 154, 157–164.
- Archibald, J.M. The Puzzle of Plastid Evolution. Curr. Biol. 2009, 19, 81–88.
- Schopf, J.W. The paleobiological record of photosynthesis. Photosynth. Res. 2011, 107, 87–101.
- Hamilton, T.L.; Bryant, D.A.; Macalady, J.L. The role of biology in planetary evolution: Cyanobacterial primary production in low-oxygen Proterozoic oceans. Environ. Microbiol. 2016, 18, 325–340.
- West, J.B. The strange history of atmospheric oxygen. Physiol. Rep. 2022, 10, e15214.
- Rasmussen, B.; Fletcher, I.R.; Brocks, J.J.; Kilburn, M.R. Reassessing the first appearance of eukaryotes and cyanobacteria. Nature 2008, 455, 1101–1104.
- Fisher, W.W.; Hemp, J.; Valentine, J.S. How did life survive Earth’s great oxygenation? Curr. Opin. Chem. Biol. 2016, 31, 166–178.
- Demoulin, C.F.; Lara, Y.J.; Cornet, L.; François, C.; Baurain, D.; Wilmotte, Y.; Javaux, E.J. Cyanobacteria evolution: Insight from the fossil record. Free Radic. Biol. Med. 2019, 140, 206–223.
- Schad, M.; Konhauser, K.O.; Sanchez-Baracaldo, P.; Kappler, A.; Bryce, C. How did the evolution of oxygenic photosynthesis influence the temporal and spatial development of the microbial iron cycle on ancient Earth? Free Radic. Biol. Med. 2019, 140, 154–188.
- Fourmier, G.P.; Morre, K.R.; Rangel, L.T.; Fayette, J.G.; Momper, L.; Bosak, T. The Archean origin of oxygenic photosynthesis and extant cyanobacterial lineages. Proc. R. Soc. B 2021, 288, 20210675.
- Shimakawa, G.; Matsuda, Y.; Nakajima, K.; Tamoi, M.; Shigeoka, S.; Miyake, C. Diverse strategies of O2 usage for preventing photo-oxidative damage under CO2 limitation during algal photosynthesis. Sci. Rep. 2017, 7, 41022.
- Weiss, E.L.; Fang, M.; Taton, A.; Szubin, R.; Palsson, B.O.; Mitchell, B.G.; Golden, S.S. An unexpected role for leucyl aminopeptidase in UV tolerance revealed by a genome-wide fitness assessment in a model cyanobacterium. Proc. Natl. Acad. Sci. USA 2022, 119, e2211789119.
- Diaz, J.M.; Plummer, S. Production of extracellular reactive oxygen species by phytoplankton: Past and future directions. J. Plankton Res. 2018, 40, 655–666.
- Faisal, M. Chromium-resistant bacteria and cyanobacteria: Impact on Cr(VI) reduction potential and plant growth. J. Ind. Microbiol. Biotechnol. 2005, 32, 615–621.
- Houot, L.; Floutier, M.; Marteyn, B.; Michaut, M.; Picciocchi, A.; Legrain, P.; Aude, J.C.; Cassier-Chauvat, C.; Chauvat, F. Cadmium triggers an integrated reprogramming of the metabolism of Synechocystis PCC6803, under the control of the Slr1738 regulator. BMC Genom. 2007, 8, 350.
- Marteyn, B.; Sakr, S.; Farci, S.; Bedhomme, M.; Chardonnet, S.; Decottignies, P.; Lemaire, S.D.; Cassier-Chauvat, C.; Chauvat, F. The Synechocystis PCC6803 MerA-like enzyme operates in the reduction of both mercury and uranium under the control of the glutaredoxin 1 enzyme. J. Bacteriol. 2013, 195, 4138–4145.
- Garcia-Calleja, J.; Cossart, T.; Pedrero, Z.; Santos, J.P.; Ouerdane, L.; Tessier, E.; Slaveykova, V.I.; Arnoux, D. Determination of the Intracellular Complexation of Inorganic and Methylmercury in Cyanobacterium Synechocystis sp. PCC 6803. Environ. Sci. Technol. 2021, 55, 13971–13979.
- Ali, S.; Mir, R.A.; Tyagi, A.; Manzar, N.; Kashyap, A.S.; Mushtaq, M.; Raina, A.; Park, S.; Sharma, S.; Mir, Z.A.; et al. Chromium Toxicity in Plants: Signaling, Mitigation, and Future Perspectives. Plants 2023, 12, 1502.
- Waldron, K.J.; Robinson, N.J. How do bacterial cells ensure thatmetalloproteins get the correct metal. Nat. Rev. Microbiol. 2009, 7, 25–35.
- Collado-Lopez, S.; Betanzos-Robledo, L.; Tellez-Rojo, M.M.; Lamadrid-Figueroa, H.; Reyes, M.; Rios, C.; Cantoral, A. Heavy Metals in Unprocessed or Minimally Processed Foods Consumed by Humans Worldwide: A Scoping Review. Int. J. Environ. Res. Public Health 2022, 19, 8651.
- Wang, R.; Sang, P.; Guo, Y.; Jin, P.; Cheng, Y.; Yu, H.; Xie, Y.; Yao, W.; Qian, H. Cadmium in food: Source, distribution and removal. Food Chem. 2023, 405, 134666.
- Fahey, R.C.; Buschbacher, R.M.; Newton, G.L. The evolution of glutathione metabolism in phototrophic microorganisms. J. Mol. Evol. 1987, 25, 81–88.
- Copley, S.D.; Dhillon, J. Lateral gene transfer and parallel evolution in the history of glutathione biosynthesis genes. Genome Biol. 2002, 3, research0025.
- Narainsamy, K.; Farci, S.; Braun, E.; Junot, C.; Cassier-Chauvat, C.; Chauvat, F. Oxidative-stress detoxification and signalling in cyanobacteria: The crucial glutathione synthesis pathway supports the production of ergothioneine and ophthalmate. Mol. Microbiol. 2016, 100, 15–24.
- Kammerscheit, X.; Chauvat, F.; Cassier-Chauvat, C. From cyanobacteria to human, MAPEG-type glutathione-S-transferases operate in cell tolerance to heat, cold, and lipid peroxidation. Front. Microbiol. 2019, 10, 2248.
- Hasanuzzaman, M.; Nahar, K.; Anee, T.I.; Fujita, M. Glutathione in plants: Biosynthesis and physiological role in environmental stress tolerance. Physiol. Mol. Biol. Plants 2017, 23, 249–268.
- Hayes, J.D.; Flanagan, J.U.; Jowsey, I.R. Glutathione transferases. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 51–88.
- Picciocchi, A.; Saguez, C.; Boussac, A.; Cassier-Chauvat, C.; Chauvat, F. CGFS-type monothiol glutaredoxins from the cyanobacterium Synechocystis PCC6803 and other evolutionary distant model organisms possess a glutathione-ligated cluster. Biochemistry 2007, 46, 15018–15026.
- Veaudor, T.; Blanc-Garin, V.; Chenebault, C.; Diaz-Santos, E.; Sassi, J.F.; Cassier-Chauvat, C.; Chauvat, F. Recent advances in the photoautotrophic metabolism of cyanobacteria: Biotechnological implications. Life 2020, 10, 71.
- Partensky, F.; Hess, W.R.; Vaulot, D. Prochlorococcus, a Marine Photosynthetic Prokaryote of Global Significance. Microbiol. Mol. Biol. Rev. 1999, 63, 106–127.
- Zwirglmaier, K.; Jardillier, L.; Ostrowski, M.; Mazard, S.; Garczarek, L.; Vaulot, D.; Not, F.; Massana, R.; Ulloa, O.; Scanlan, D.J. Global phylogeography of marine Synechococcus and Prochlorococcus reveals a distinct partitioning of lineages among oceanic biomes. Environ. Microbiol. 2008, 10, 147–161.
- Dai, W.; Chen, M.; Myers, C.; Ludtke, S.J.; Pettitt, B.M.; King, J.A.; Schmid, M.F.; Chiu, W. Visualizing Individual RuBisCO and Its Assembly into Carboxysomes in Marine Cyanobacteria by Cryo-Electron Tomography. J. Mol. Biol. 2018, 430, 4156–4167.
- Nelson, C.; Garcia-Pichel, F. Beneficial Cyanosphere Heterotrophs Accelerate Establishment of Cyanobacterial Biocrust. Appl. Environ. Microbiol. 2021, 87, e0123621.
- Singh, J.S.; Kumar, A.; Rai, A.N.; Singh, D.P. Cyanobacteria: A precious bio-resource in agriculture, ecosystem, and environmental sustainability. Front. Microbiol. 2016, 7, 529.
- Panjiar, N.; Mishra, S.; Yadav, A.N.; Verma, P. Functional Foods from Cyanobacteria. In Microbial Functional Foods and Nutraceuticals; Gupta, V.K., Treichel, H., Shapaval, V., de Oliveira, L.A., Tuohy, M.G., Eds.; Wiley: Hoboken, NY, USA, 2017.
- Sheha, G.R.; Yadav, R.K.; Chatrath, A.; Gerard, M.; Tripathi, K.; Govindsamy, V.; Abraham, G. Perspectives on the potential application of cyanobacteria in the alleviation of drought and salinity stress in crop plants. J. Appl. Phycol. 2021, 33, 3761–3778.
- Liu, D.; Liberton, M.; Hendry, J.I.; Aminian-Dehkordi, J.; Maranas, C.D.; Pakrasi, H.B. Engineering biology approaches for food and nutrient production by cyanobacteria. Curr. Opin. Biotechnol. 2021, 67, 1–6.
- Cassier-Chauvat, C.; Dive, V.; Chauvat, F. Cyanobacteria: Photosynthetic factories combining biodiversity, radiation resistance, and genetics to facilitate drug discovery. Appl. Microbiol. Biotechnol. 2017, 101, 1359–1364.
- Demay, J.; Bernard, C.; Reinhardt, A.; Marie, B. Natural products from cyanobacteria: Focus on beneficial activities. Mar. Drugs 2019, 17, 320.
- Kirsch, F.; Klähn, S.; Hagemann, M. Salt-Regulated Accumulation of the Compatible Solutes Sucrose and Glucosylglycerol in Cyanobacteria and Its Biotechnological Potential. Front. Microbiol. 2019, 10, 2139.
- Pade, N.; Hagemann, M. Salt Acclimation of Cyanobacteria and Their Application in Biotechnology. Life 2015, 5, 25–49.
- Kollmen, J.; Strieth, D. The Beneficial Effects of Cyanobacterial Co-Culture on Plant Growth. Life 2022, 12, 223.
- Cassier-Chauvat, C.; Chauvat, F. Cell division in cyanobacteria. In The Cell Biology of Cyanobacteria; Flores, E., Herrero, A., Eds.; Caister Academic Press: Poole, UK, 2014.
- Herrero, A.; Stavans, J.; Flores, E. The multicellular nature of filamentous heterocyst-forming cyanobacteria. FEMS Microbiol. Rev. 2016, 40, 831–854.
- Cassier-Chauvat, C.; Blanc-Garin, V.; Chauvat, F. Genetic, Genomics, and Responses to Stresses in Cyanobacteria: Biotechnological Implications. Genes 2021, 12, 500.
- Cassier-Chauvat, C.; Veaudor, T.; Chauvat, F. Comparative genomics of DNA recombination and repair in cyanobacteria: Biotechnological implications. Front. Microbiol. 2016, 7, 1809.
- Meister, A. On the discovery of glutathione. Trends Biochem. Sci. 1988, 13, 185–188.
- Noctor, G.; Mhamdi, A.; Chaouch, S.; Han, Y.; Neukermans, J.; Marquez-Garcia, B.; Queval, G.; Foyer, C.H. Glutathione in plants: An integrated overview. Plant Cell Environ. 2012, 35, 454–484.
- Berndt, C.; Lillig, C.H. Glutathione, Glutaredoxins, and Iron. Antioxid. Redox Signal. 2017, 27, 1235–1251.
- Scire, A.; Cianfruglia, L.; Minnelli, C.; Bartolinin, D.; Torquato, P.; Principato, G.; Galli, F.; Armeni, T. Glutathione compartmentalization and its role in glutathionylation and other regulatory processes of cellular pathways. BioFactors 2018, 45, 152–168.
- Grant, C.M.; Maciver, F.H.; Dawes, I.W. Glutathione is an essential metabolite required for resistance to oxidative stress in the yeast Saccharomyces cerevisiae. Curr. Genet. 1996, 29, 511–515.
- Spector, D.; Labarre, J.; Toledano, M.B. A genetic investigation of the essential role of glutathione: Mutations in the proline biosynthesis pathway are the only suppressors of glutathione auxotrophy in yeast. J. Biol. Chem. 2001, 276, 7011–7016.
- Shi, Z.-Z.; Osei-Frimpong, J.; Kala, G.; Lieberman, M.W. Glutathione synthesis is essential for mouse development but not for cell growth in culture. Proc. Natl. Acad. Sci. USA 2000, 97, 5101–5106.
- Cairns, N.G.; Pasternak, M.; Wachter, A.; Cobbett, C.S.; Meyer, A.J. Maturation of Arabidopsis seeds is dependent on glutathione biosynthesis within the embryo. Plant Physiol. 2006, 141, 446–455.
- Greenberg, J.T.; Demple, B. Glutathione in Escherichia coli is dispensable for resistance to H2O2 and gamma radiation. J. Bacteriol. 1986, 168, 1026–1029.
- Fuchs, J.A.; Warner, H.R. Isolation of an Escherichia coli mutant deficient in glutathione synthesis. J. Bacteriol. 1975, 124, 140–148.
- Daws, T.; Lim, C.J.; Fuchs, J.A. In vitro construction of gshB::kan in Escherichia coli and use of gshB::kan in mapping the gshB locus. J. Bacteriol. 1989, 171, 5218–5221.
- Chesney, J.A.; Eaton, J.W.; Mahoney, J.R. Bacterial Glutathione: A Sacrificial Defense against Chlorine Compounds. J. Bacteriol. 1996, 178, 2131–2135.
- Howden, R.; Andersen, C.R.; Coldsbrough, P.; Cobbett, C.S. A cadmium-sensitive, glutathione-deficient mutant of Arabidopsis thaliana. PLANT Physiol. 1995, 107, 1067–1073.
- Shanmugam, V.; Tsednee, L.; Yeh, K.-C. Zinc Tolerance Induced by Iron 1 reveals the importance of glutathione in the cross-homeostasis between zinc and iron in Arabidopsis thaliana. Plant J. 2012, 69, 1006–1017.
- Ponce-Toledo, R.I.; Deschamps, P.; López-García, P.; Zivanovic, Y.; Benzerara, K.; Moreira, D. An Early-Branching Freshwater Cyanobacterium at the Origin of Plastids. Curr. Biol. 2017, 27, 386–391.
- Pinevich, A.V. Chloroplast history clarified by the criterion of light-harvesting complex. Biosystem 2020, 196, 104176.
- Seregin, I.V.; Kozhevnikova, A.D. Phytochelatins: Sulfur-Containing Metal(loid)-Chelating Ligands in Plants. Int. J. Mol. Sci. 2023, 24, 2430.
- Yang, J.; Li, W.; Wang, D.; Wu, H.; Li, Z.; Ye, Q. Characterization of bifunctional L-glutathione synthetases from Actinobacillus pleuropneumoniae and Actinobacillus succinogenes for efficient glutathione biosynthesis. Appl. Microbiol. Biotechnol. 2016, 100, 6279–6289.
- Gopal, S.; Borovok, A.; Ofer, M.; Yanku, G.; Cohen, W.; Kreft, J.; Aharonowitz, Y. A multidomain fusion protein in Listeria monocytogenes catalyzes the two primary activities for glutathione biosynthesis. J. Bacteriol. 2005, 187, 3839–3847.
- Vergauwen, B.; De Vos, D.; Van Beeumen, J.J. Characterization of the bifunctional gamma-glutamate-cysteine ligase/glutathione synthetase (GshF) of Pasteurella multocida. J. Biol. Chem. 2006, 281, 4380–4394.
- Janowiak, B.; Griffith, O.W. Glutathione synthesis in Streptococcus agalactiae One protein accounts for gamma-glutamylcysteine synthetase and glutathione synthetase activities. J. Biol. Chem. 2005, 280, 11829–11839.
- Li, W.; Li, Z.; Yang, J.; Ye, Q. Production of glutathione using a bifunctional enzyme encoded by gshF from Streptococcus thermophilus expressed in Escherichia coli. J. Bacteriol. 2011, 154, 261–268.
- Santos, L.O.; Silva, P.G.P.; Lemos, W.J.F.L.; de Oliveira, V.S.; Anschau, A. Glutathione production by Saccharomyces cerevisiae: Current state and perspectives. Appl. Environ. Microbiol. 2022, 106, 1879–1894.
- Jeon, G.-B.; Lee, H.-J.; Park, J.P.; Park, K.; Choi, C.-H.; Kim, S.-K. Efficient production of glutathione in Saccharomyces cerevisiae via a synthetic isozyme system. Biotechnol. J. 2022, 8, 2200398.
- Couto, N.; Wood, J.; Barber, J. The role of glutathione reductase and related enzymes on cellular redox homoeostasis network. Free Radic. Biol. Med. 2016, 95, 27–42.
- Ali, V.; Behera, S.; Nawaz, A.; Equbal, A.; Pandey, K. Unique thiol metabolism in trypanosomatids: Redox homeostasis and drug resistance. Adv. Parasitol. 2022, 117, 75–155.
- Chandrangsu, P.; Van Loi, W.; Antelman, H.; Helman, J.D. The Role of Bacillithiol in Gram-Positive Firmicutes. Antioxid. Redox Signal. 2018, 28, 445–462.
- Sao Emani, C.; Gallant, J.L.; Wild, I.J.; Baker, B. The role of low molecular weight thiols in Mycobacterium tuberculosis. Tuberculosis 2019, 116, 44–55.
- Qiu, Y.; Chen, Z.; Su, E.; Wang, L.; Sun, L.; Lei, P.; Xu, H.; Li, S. Recent Strategies for the Biosynthesis of Ergothioneine. J. Agric. Food Chem. 2021, 69, 13682–13690.
- Cumming, B.M.; Chinta, K.C.; Reddy, V.P.; Steyn, A.J.C. Role of Ergothioneine in Microbial Physiology and Pathogenesis. Antioxid. Redox Signal. 2018, 28, 431–444.
- Borodina, I.; Kenny, L.C.; McCarthy, C.M.; Paramasivan, K.; Pretorius, E.; Roberts, T.J.; van der Hoek, S.A.; Kell, D.B. The biology of ergothioneine, an antioxidant nutraceutical. Nutr. Res. Rev. 2020, 33, 190–217.
- Nakamichi, N.; Tsuzuku, S.; Shibagaki, F. Ergothioneine and central nervous system diseases. Neurochem. Res. 2022, 47, 2513–2521.
- Yadan, J.-C. Matching chemical properties to molecular biological activities opens a new perspective on l-ergothioneine. FEBS Lett. 2022, 596, 1299–1312.
- Liu, H.-M.; Tang, W.; Wang, X.-Y.; Jiang, J.-J.; Zhang, W.; Wang, W. Safe and Effective Antioxidant: The Biological Mechanism and Potential Pathways of Ergothioneine in the Skin. Molecules 2023, 28, 1648.
- Fahey, R.C. Novel Thiols of Prokaryotes. Annu. Rev. Microbiol. 2001, 55, 333–356.
- Stampfli, A.R.; Blankenfeldt, W.; Seebeck, F.P. Structural basis of ergothioneine biosynthesis. Curr. Opin. Struct. Biol. 2020, 65, 1–8.
- Gokçe, G.; Zuhuri, M.; Erfuna, E. Ergothioneine prevents endothelial dysfunction induced by mercury chloride. Exp. Ther. Med. 2018, 15, 4697–4702.
- Waley, S.G. Acidic peptides of the lens. 3. The structure of ophthalmic acid. Biochem. J. 1958, 68, 189–192.
- Tsuboii, S.; Hirota, K.; Ogata, K.; Ohmori, S. Ophthalmic and Norophthalmic Acid in Lens, Liver, and Brain of Higher Animals. Anal. Biochem. 1984, 136, 520–524.
- Soga, T.; Baran, R.; Suematsu, M.; Ueno, Y.; Ikeda, S.; Sakurakawa, T.; Kakazu, Y.; Ishikawa, T.; Robert, M.; Nishioka, T.; et al. Differential metabolomics reveals ophthalmic acid as an oxidative stress biomarker indicating hepatic glutathione consumption. J. Biol. Chem. 2006, 281, 16768–16776.
- Dello, S.A.W.G.; Neis, E.P.J.G.; de Jong, M.C.; van Eijk, H.M.H.; Kicken, C.H.; Olde Damink, S.W.M.; Dejong, C.H.C. Systematic review of ophthalmate as a novel biomarker of hepatic glutathione depletion. Clin. Nutr. 2013, 32, 325–330.
- Servillo, L.; Castaldo, D.; Giovane, A.; Casale, R.; D’Onofrio, N.; Cautela, D.; Balestrieri, M.L. Ophthalmic acid is a marker of oxidative stress in plants as in animals. Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 991–998.
- Sajiki, K.; Pluskai, T.; Shimanuki, M.; Yanagida, M. Metabolomic analysis of fission yeast at the onset of nitrogen starvation. Metabolites 2013, 3, 1118–1129.
- Ito, T.; Yamauchi, A.; Hemmi, H.; Yoshimura, T. Ophthalmic acid accumulation in an Escherichia coli mutant lacking the conserved pyridoxal 5′-phosphate-binding protein YggS. J. Biosci. Bioeng. 2016, 122, 689–693.
- Ito, T.; Ohkama-Ohtsu, N. Degradation of glutathione and glutathione conjugates in plants. J. Exp. Bot. 2023, erad018.
- Saini, M.; Kashyap, A.; Bindal, S.; Saini, K.; Gupta, R. Bacterial Gamma-Glutamyl Transpeptidase, an Emerging Biocatalyst: Insights into Structure–Function Relationship and Its Biotechnological Applications. Front. Microbiol. 2021, 12, 641251.
- Yang, J.; Bai, W.; Zeng, X.; Cui, C. Gamma glutamyl peptides: The food source, enzymatic synthesis, kokumi-active and the potential functional properties—A review. Trends Food Sci. Technol. 2019, 91, 339–346.
- Lee, D.S.; Evans, J.C.; Robins, S.J.; Wilson, P.W.; Albano, I.; Fox, C.S.; Wang, T.J.; Benjamin, E.J.; D’Agostino, R.B.; Vasan, R.S. Gamma Glutamyl Transferase and Metabolic Syndrome, Cardiovascular Disease, and Mortality Risk. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 127–133.
- Schulz, G.; Schirmer, S.H.; Sachsenheimer, W.; Pai, E.F. The structure of the flavoenzyme glutathione reductase. Nature 1978, 273, 120–124.
- Scrutton, N.S.; Berry, A.; Perham, R.N. Redesign of the coenzyme specificity of a dehydrogenase by protein engineering. Nature 1990, 343, 38–43.
- Perham, R.N.; Scrutton, N.S.; Berry, A. New enzymes for old: Redesigning the coenzyme and substrate specificities of glutathione reductase. Bioessays 1991, 10, 515–525.
- Lin, T.H.; Rao, M.Y.; Lu, H.W.; Chiou, C.W.; Lin, S.T.; Chao, H.W.; Zheng, Z.L.; Cheng, H.C.; Lee, T.M. A role for glutathione reductase and glutathione in the tolerance of Chlamydomonas reinhardtii to photo-oxidative stress. Physiol. Plant. 2018, 162, 35–48.
- Davis, N.K.; Greer, S.; Jones-Mortimer, M.C.; Perham, R.N. Isolation and mapping of glutathione reductase-negative mutants of Escherichia coli K12. J. Gen Microbiol. 1982, 128, 1631–1634.
- Tuggle, C.K.; Fuchs, J.A. Glutathione reductase is not required for maintenance of reduced glutathione in Escherichia coli K-12. J. Bacteriol. 1985, 162, 448–450.
- Alonso-Moraga, A.; Bocanegra, A.; Torres, J.M.; Pueyo, C. Glutathione status and sensitivity to GSH-reacting compounds of Escherichia coli strains deficient in glutathione metabolism and/or catalase activity. Mol. Cell. Biochem. 1987, 73, 61–68.
- Kunert, K.J.; Cresswell, C.F.; Schmidt, A.; Mullineaux, P.M.; Foyer, C.H. Variations in the activity of glutathione reductase and the cellular glutathione content in relation to sensitivity to methylviologen in Escherichia coli. Arch. Biochem. Biophys. 1990, 282, 233–238.
- Barbado, C.; Ramirez, M.; Blanco, M.A.; Lopez-Barea, J.; Pueyo, C. Mutants of Escherichia coli sensitive to hydrogen peroxide. Curr. Microbiol. 1983, 8, 251–253.
- Collison, L.P.; Dawes, I.W. Isolation, characterization and overexpression of the yeast gene, GLRI, encoding glutathione reductase. Gene 1995, 156, 123–127.
- Muller, E.G. A glutathione reductase mutant of yeast accumulates high levels of oxidized glutathione and requires thioredoxin for growth. Mol. Biol. Cell 1996, 7, 1805–1813.
- Grant, C.M.; Collison, L.P.; Roe, J.H.; Dawes, I.W. Yeast glutathione reductase is required for protection against oxidative stress and is a target gene for yAP-1 transcriptional regulation. Mol. Microbiol. 1996, 21, 171–179.
- Saing, T.; Lagman, M.; Castrillon, J.; Gutierrez, E.; Guilford, F.T.; Venketaraman, V. Analysis of glutathione levels in the brain tissue samples from HIV-1-positive individuals and subject with Alzheimer’s disease and its implication in the pathophysiology of the disease process. Biochim. Biophys. Acta Clin. 2016, 29, 38–44.
- Yan, J.; Meng, X.; Wancket, L.M.; Lintner, K.; Nelin, L.D.; Chen, B.; Francis, K.P.; Smith, C.V.; Rogers, L.K.; Liu, Y. Glutathione reductase facilitates host defense by sustaining phagocytic oxidative burst and promoting the development of neutrophil extracellular traps. J. Immunol. 2012, 1888, 2316–2327.
- Berndt, C.; Lillig, C.H.; Flohé, L. Redox regulation by glutathione needs enzymes. Front. Pharmacol. 2014, 5, 168.
- Josephy, P.D. Genetic variations in human glutathione transferase enzymes: Significance for pharmacology and toxicology. Hum. Genom. Proteom. 2010, 2010, 876940.
- Carvalho, A.N.; Marques, C.; Guedes, R.C.; Castro-Caldas, M.; Rodrigues, E.; Van Horssen, J.; Gama, M.J. S-Glutathionylation of Keap1: A new role for glutathione S-transferase pi in neuronal protection. FEBS Lett. 2016, 590, 1455–1466.
- Allocati, N.; Masulli, M.; Di Ilio, C.; Federici, L. Glutathione transferases: Substrates, inihibitors and pro-drugs in cancer and neurodegenerative diseases. Oncogenesis 2018, 7, 8.
- Bocedi, A.; Noce, A.; Marrone, G.; Noce, G.; Cattani, G.; Gambardella, G.; Di Lauro, M.; Di Daniele, N.; Ricci, G. Glutathione Transferase P1-1 an Enzyme Useful in Biomedicine and as Biomarker in Clinical Practice and in Environmental Pollution. Nutrients 2019, 11, 1741.
- Sylvestre-Gonon, E.; Law, S.R.; Schwartz, M.; Robe, K.; Keech, O.; Didierjean, C.; Dubos, C.; Rouhier, N.; Hecker, A. Functional, structural and biochemical features of plant serinyl-glutathione transferases. Front. Plant Sci. 2019, 10, 608.
- Kumar, S.; Trivedi, P.K. Glutathione S-Transferases: Role in Combating Abiotic Stresses Including Arsenic Detoxification in Plants. Front. Plant Sci. 2018, 9, 751.
- Schwartz, M.; Perrot, T.; Aubert, E.; Dumarçay, S.; Favier, F.; Gérardin, P.; Morel-Rouhier, M.; Mulliert, G.; Saiag, F.; Didierjean, C.; et al. Molecular recognition of wood polyphenols by phase II detoxification enzymes of the white rot Trametes versicolor. Sci. Rep. 2018, 8, 8472.
- Gullner, G.; Komives, T.; Kiraly, L.; Schröder, P. Glutathione S-Transferase Enzymes in Plant-Pathogen Interactions. Front. Plant Sci. 2018, 9, 1836.
- Kammerscheit, X.; Hecker, A.; Rouhier, N.; Chauvat, F.; Cassier-Chauvat, C. Methylglyoxal Detoxification Revisited: Role of Glutathione Transferase in Model Cyanobacterium Synechocystis sp. Strain PCC 6803. MBio 2020, 11.
- Allocati, N.; Federici, L.; Masulli, M.; Di Ilio, C. Glutathione transferases in bacteria. FEBS J. 2009, 276, 58–75.
- Pandey, T.; Chhetri, G.; Chinta, R.; Kumar, B.; Singh, D.B.; Tripathi, T.; Singh, A.K. Functional classification and biochemical characterization of a novel rho class glutathione S-transferase in Synechocystis PCC 6803. FEBS Open Bio 2015, 5, 1–7.
- Rani, R.; Simarani, K.; Alias, Z. Functional Role of Beta Class Glutathione Transferases and Its Biotechnological Potential (Review). Biochemistry 2022, 49, S20–S29.
- Lederer, B.; Böger, P. A ligand function of glutathione S-transferase. Z. fur Naturforsch. Sect. C J. Biosci. 2005, 60, 166–171.
- Oakley, A. Glutathione transferases: A structural perspective. Drug Metab. Rev. 2011, 43, 138–151.
- Mocchetti, E.; Morette, L.; Mulliert, G.; Mathiot, S.; Guillot, B.; Dehez, F.; Chauvat, F.; Cassier-Chauvat, C.; Brochier-Armanet, C.; Didierjean, C.; et al. Biochemical and Structural Characterization of Chi-Class Glutathione Transferases: A Snapshot on the Glutathione Transferase Encoded by sll0067 Gene in the Cyanobacterium Synechocystis sp. Strain PCC 6803. Biomolecules 2022, 12, 1466.
- Thornalley, P.J. Protein and nucleotide damage by glyoxal and methylglyoxal in physiological systems-role in ageing and disease. Drug Metab. Drug Interact. 2008, 23, 125–150.
- Lee, C.; Park, C. Bacterial responses to glyoxal and methylglyoxal: Reactive electrophilic species. Int. J. Mol. Sci. 2017, 18, 169.
- Mostofa, M.G.; Ghosh, A.; Li, Z.G.; Siddiqui, M.N.; Fujita, M.; Tran, L.S.P. Methylglyoxal—A signaling molecule in plant abiotic stress responses. Free Radic. Biol. Med. 2018, 22, 96–109.
- Schalkwijk, C.G.; Stehouwer, C.D.A. Methylglyoxal, a highly reactive dicarbonyl compound, in diabetes, its vascular complications, and other age-related diseases. Physiol. Rev. 2020, 100, 407–461.
- de Bari, L.; Scirè, A.; Minnelli, C.; Cianfruglia, L.; Kalapos, M.P.; Armeni, T. Interplay among oxidative stress, methylglyoxal pathway and s-glutathionylation. Antioxidants 2021, 10, 19.
- Ramachandra Bhat, L.; Vedantham, S.; Krishnan, U.M.; Rayappan, J.B.B. Methylglyoxal—An emerging biomarker for diabetes mellitus diagnosis and its detection methods. Biosens. Bioelectron. 2019, 133, 107–124.
- Kalapos, M.P. Methylglyoxal in living organisms: Chemistry, biochemistry, toxicology and biological implications. Toxicol. Lett. 1999, 110, 145–175.
- He, Y.; Zhou, C.; Huang, M.; Tang, C.; Liu, X.; Yue, Y.; Diao, Q.; Zheng, Z.; Liu, D. Glyoxalase system: A systematic review of its biological activity, related-diseases, screening methods and small molecule regulators. Biomed. Pharmacother. 2020, 131, 110663.
- Rabbani, N.; Al-Motawa, M.; Thornalley, P.J. Protein glycation in plants—An under-researched field with much still to discover. Int. J. Mol. Sci. 2020, 21, 3942.
- Nemet, I.; Varga-Defterdarovic, L. Methylglyoxal-derived beta-carbolines formed from tryptophan and its derivates in the Maillard reaction. Amino Acids 2007, 32, 291–293.
- Eggen, M.D.; Glomb, M.A. Analysis of Glyoxal- and Methylglyoxal-Derived Advanced Glycation End Products during Grilling of Porcine Meat. J. Agric. Food Chem. 2021, 69, 15374–15383.
- Kaur, C.; Sharma, S.; Hasan, M.R.; Pareek, A.; Singla-Pareek, S.L.; Sopory, S.K. Characteristic variations and similarities in biochemical, molecular, and functional properties of glyoxalases across prokaryotes and eukaryotes. Int. J. Mol. Sci. 2017, 18, 250.
- Van Loi, V.; Rossius, M.; Antelmann, H. Redox regulation by reversible protein S-thiolation in bacteria. Front. Microbiol. 2015, 6, 147.
- Muller-Schussele, S.J.; Bohle, F.; Rossi, J.; Trost, P.; Meyer, A.; Zaffagnini, M. Plasticity in plastid redox networks: Evolution of glutathione-dependent redox cascades and glutathionylation sites. BMC Plant Biol. 2021, 21, 322.
- Mondal, S.; Kumar, V.; Singh, S.P. Phylogenetic distribution and structural analyses of cyanobacterial glutaredoxins (Grxs). Comput. Biol. Chem. 2020, 84, 107141.
- Holmgren, A. Hydrogen donor system for Escherichia coli ribonucleoside diphosphate reductase dependent upon glutathione. Proc. Natl. Acad. Sci. USA 1976, 73, 2275–2279.
- Laurent, T.C.; Moore, E.C.; Reichard, P. Enzymatic Synthesis of Deoxyribonucleotides. Iv. Isolation and and characterization of thioredoxin, the hydrogen donor from Escherichia coli B. J. Biol. Chem. 1964, 239, 3436–3444.
- Martin, J.L. Thioredoxin—A fold for all reasons. Curr. Biol. 1995, 3, 45–250.
- Trnka, D.; Engelke, A.D.; Gellert, M.; Moseler, A.; Hossain, M.F.; Lindenberg, T.T.; Pedroletti, L.; Odermatt, B.; de Souza, J.V.; Bronowska, A.K.; et al. Molecular basis for the distinct functions of redox-active and FeS-transfering glutaredoxins. Nat. Commun. 2020, 11, 3445.
- Hider, R.C.; Kong, X.L. Glutathione: A key component of the cytoplasmic labile iron pool. BioMetals 2011, 24, 1179–1187.
- Kumar, C.; Igbaria, A.; D’Autreaux, B.; Planson, A.-G.; Junot, C.; Godat, E.; Bacchawat, A.K.; Delaunay-Moisan, A.; Toledano, M.B. Glutathione revisited: A vital function in iron metabolism and ancillary role in thiol-redox control. EMBO J. 2011, 30, 2044–2056.
- Xu, L.; Liu, Y.; Chen, X.; Zhong, H.; Wang, Y. Ferroptosis in life: To be or not to be. Biomed. Pharmacol. 2023, 159, 114241.
- Aguilera, A.; Berdun, F.; Bartoli, C.; Steelheart, C.; Alegre, M.; Bayir, H.; Tyurina, Y.Y.; Kagan, V.E.; Salerno, G.; Pagnussat, G.; et al. C-ferroptosis is an iron-dependent form of regulated cell death in cyanobacteria. J. Cell Biol. 2022, 221, e201911005.
- Conrad, M.; Kagan, V.E.; Bayir, H.; Pagnussat, G.C.; Head, B.; Traber, M.G.; Stockwell, B.R. Regulation of lipid peroxidation and ferroptosis in diverse species. Genes Dev. 2018, 32, 602–619.
- Lobus, N.V.; Kulikovskly, M.S. The Co-Evolution Aspects of the Biogeochemical Role of Phytoplankton in Aquatic Ecosystems: A Review. Biology 2023, 12, 92.
- Saito, M.A.; Sigman, D.M.; Morel, F.M.M. The bioinorganic chemistry of the ancient ocean: The co-evolution of cyanobacterial metal requirements and biogeochemical cycles at the Archean–Proterozoic boundary? Inorg. Chim. Acta 2003, 356, 308–318.
- Qiu, G.-W.; Koedooder, C.; Qiu, B.-S.; Shaked, Y.; Keren, N. Iron transport in cyanobacteria—From molecules to communities. Trends Microbiol. 2022, 30, 229–240.
- Lill, R. Function and biogenesis of iron–sulphur proteins. Nature 2009, 460, 831–838.
- Przybyla-Toscano, J.; Roland, M.; Gaymard, F.; Couturier, J.; Rouhier, N. Roles and maturation of iron–sulfur proteins in plastids. J. Biol. Inorg. Chem. 2018, 23, 545–566.
- de Bont, L.; Donnay, N.; Couturier, J.; Rouhier, N. Redox regulation of enzymes involved in sulfate assimilation and in the synthesis of sulfur-containing amino acids and glutathione in plants. Front. Plant Sci. 2022, 13, 958490.
- Sipos, K.; Lange, H.; Fekete, Z.; Ullmann, P.; Lill, R.; Kispal, G. Maturation of cytosolic iron-sulfur proteins requires glutathione. J. Biol. Chem. 2002, 277, 26944–26949.
- Srour, N.; Gervason, S.; Hook, M.H.; Monfort, B.; Want, K.; Larkem, D.; Trabelsi, N.; Landrot, G.; Zitolo, A.; Fonda, E.; et al. Iron Insertion at the Assembly Site of the ISCU Scaffold Protein Is a Conserved Process Initiating Fe-S Cluster Biosynthesis. J. Am. Chem. Soc. 2022, 144, 17496–17515.
- Thompson, Z.; Fidai, I.; Wachnowsky, C.; Hendricks, A.L.; Cowan, J.A. Spectroscopic and functional characterization of the scaffold protein Nfu from Synechocystis PCC6803. Biochimie 2022, 192, 51–62.
- Lillig, C.H.; Berndt, C.; Vergnolle, O.; Lohn, M.E.; Hudemann, C.; Bill, E.; Holgren, A. Characterization of human glutaredoxin 2 as iron-sulfur protein: A possible role as redox sensor. Proc. Natl. Acad. Sci. USA 2005, 102, 8168–8173.
- Feng, Y.; Zhong, N.; Rouhier, N.; Hase, T.; Kusunoki, M.; Jacquot, J.P.; Jin, C.; Xia, B. Structural insight into poplar glutaredoxin C1 with a bridging iron-sulfur cluster at the active site. Biochemistry 2006, 45, 7996–8008.