Metabolic disorders are a spectrum of diseases that affect normal metabolic functioning and regulation. More than 500 metabolic disorders exist, two of the most common being diabetes mellitus and obesity. Obesity is a multifactorial disorder involving the interaction of genetics, lifestyle and environment. It results in excessive adipose tissue deposition and is defined by a body mass index (BMI) greater than 30 kg/m2. In addition, obesity is the leading cause of metabolic syndrome and insulin resistance. Type 2 diabetes is characterized by an elevated blood glucose level, a chronic hyperglycemic state caused by a combination of pancreatic β-cell loss through apoptosis and insulin resistance in peripheral tissues, such as skeletal muscle. By contrast, type 1 diabetes is caused by autoimmune attack upon pancreatic β-cells, causing an almost complete loss of insulin production and secretion. Whilst a genetic predisposition can underlie the onset of type 1 diabetes, with particular loci of interest having been identified, environmental factors may also contribute. Well-established risk factors for type 2 diabetes and obesity are a sedentary lifestyle, poor nutrition, insulin resistance, environmental factors and genetics.
- persistent organic pollutants
- atmospheric pollution
- obesity
- type 1 diabetes
- type 2 diabetes
- gestational diabetes
- metabolic disorders
- heavy metals
- adipogenesis
1. The Impact of Pollution on Obesity
1.1. The Development of Obesity
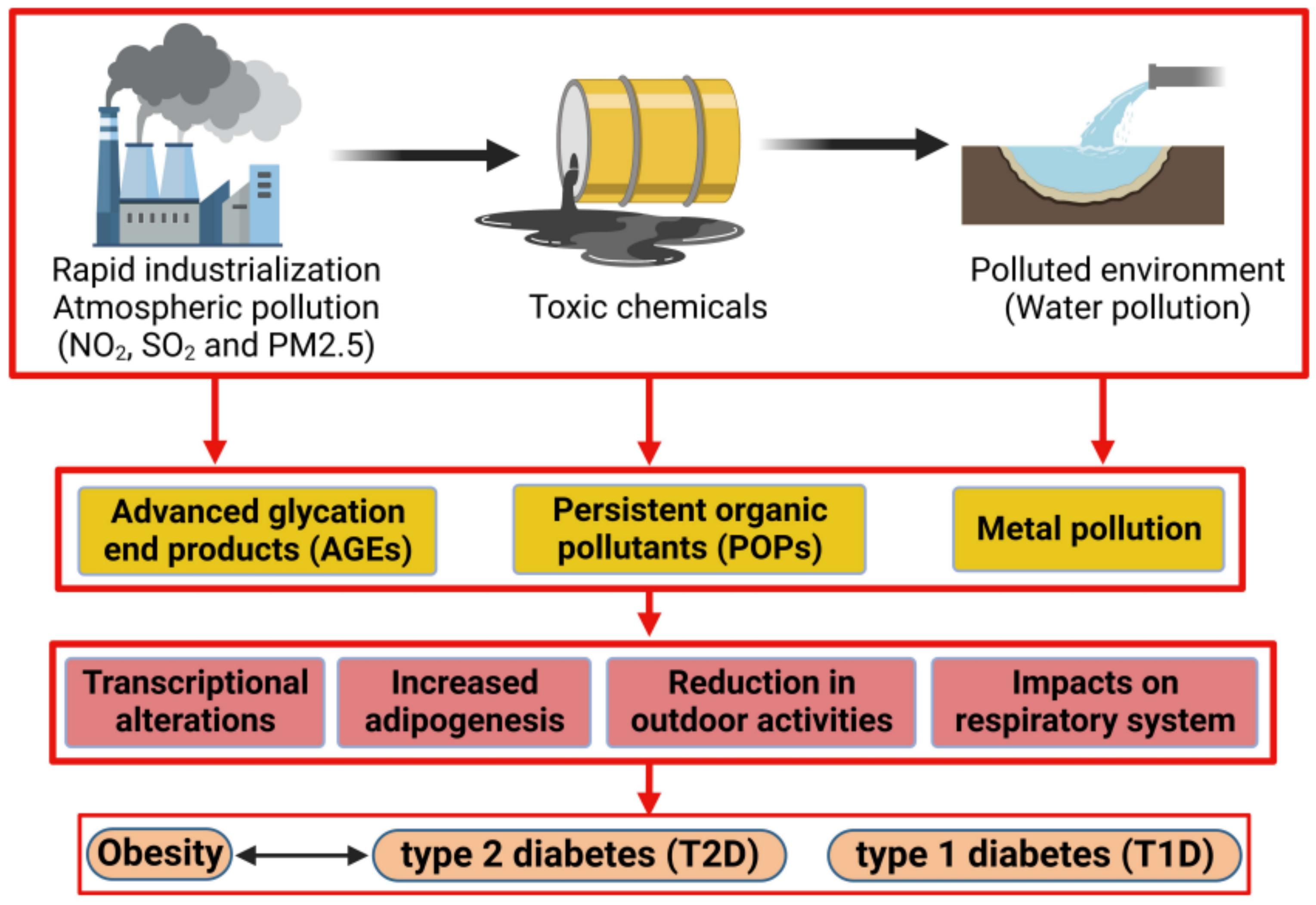
1.2. Advanced Glycation End Product (AGE) Impact on Obesity Prevalence
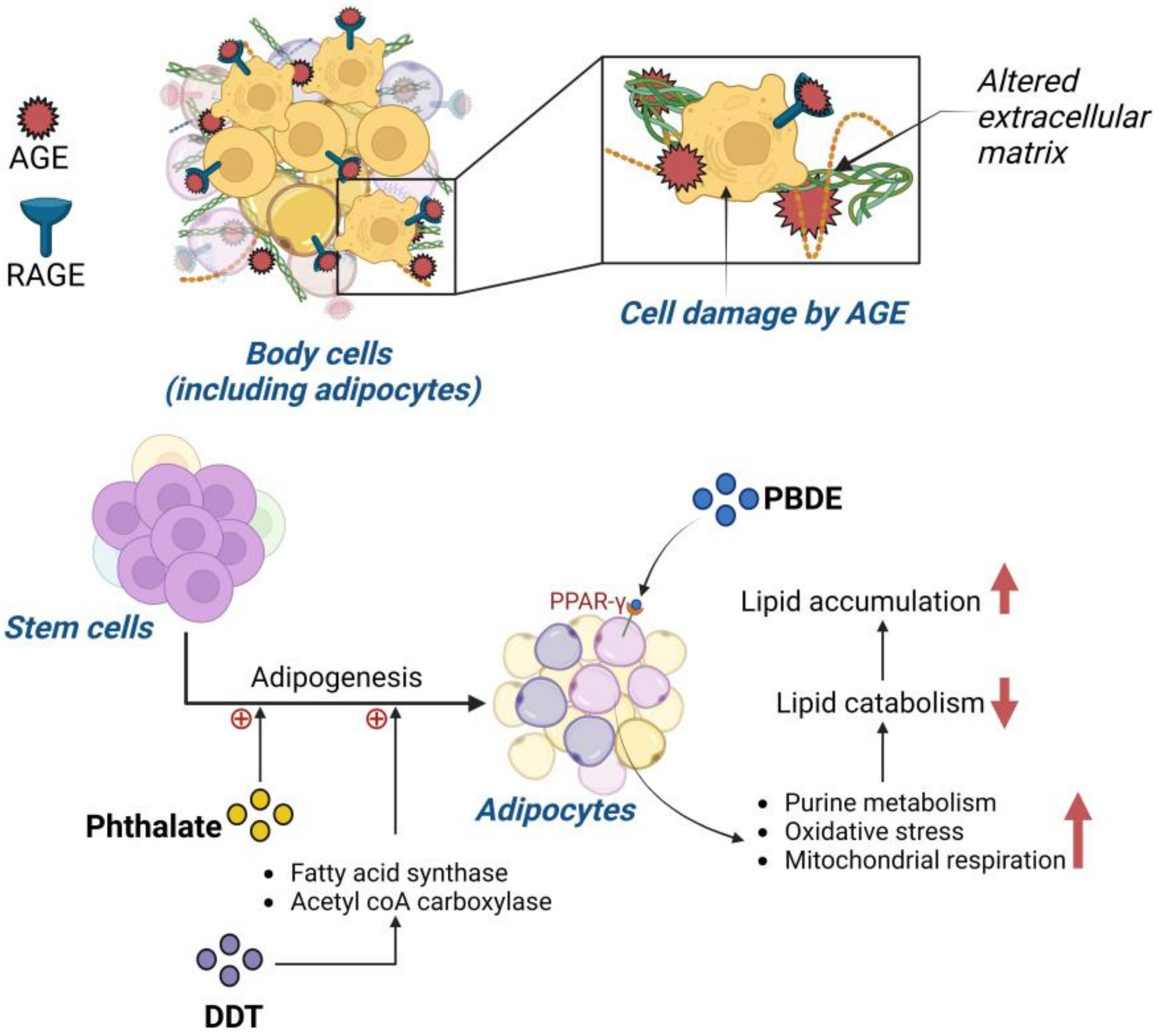
1.3. Impact of Persistent Organic Pollutants on Obesity
1.4. Pollution and the Impact on Childhood Obesity
2. Impact of Pollution on Type 2 Diabetes Mellitus (T2D)
2.1. The Development of Type 2 Diabetes Mellitus
2.2. The Role of Metals in Inducing Type 2 Diabetes Mellitus
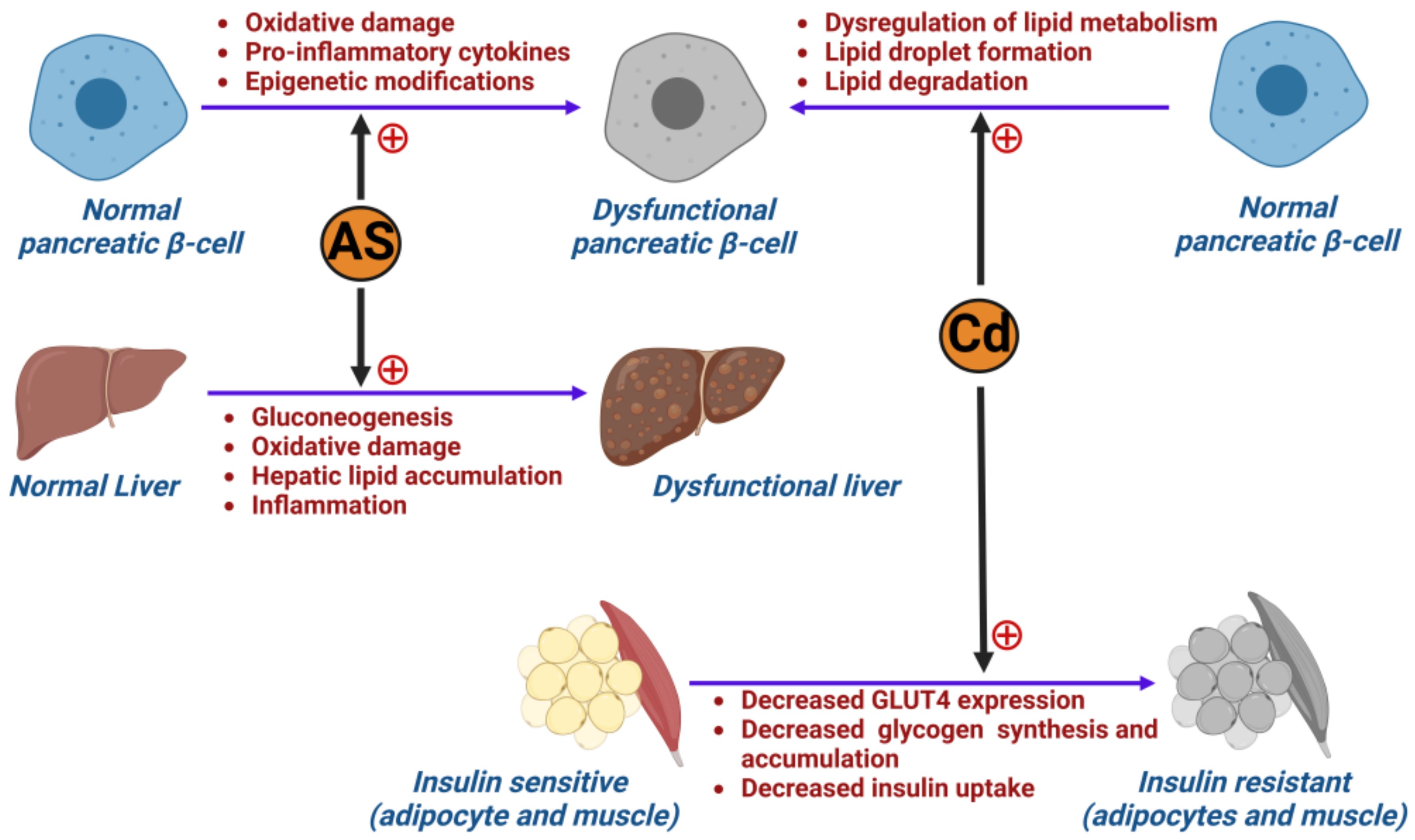
2.3. Air Pollution and Its Impact on Developing Type 2 Diabetes (T2D)
3. The Impact of Pollution on Type 1 Diabetes Mellitus (T1D)
3.1. Impact of Pollution on Pancreatic β-Islet Cells
3.2. Impact of Pollution on Childhood Type 1 Diabetes Mellitus
4. Impact of Pollution on Gestational Diabetes (GDM)
4.1. The Development of Gestational Diabetes
4.2. Association between Particulate Matter (PM) and Gestational Diabetes (GDM)
This entry is adapted from the peer-reviewed paper 10.3390/ijms24108870
References
- Seo, M.Y.; Kim, S.-H.; Park, M.J. Air pollution and childhood obesity. Clin. Exp. Pediatr. 2020, 63, 382–388.
- Xu, X.; Yavar, Z.; Verdin, M.; Ying, Z.; Mihai, G.; Kampfrath, T.; Wang, A.; Zhong, M.; Lippmann, M.; Chen, L.-C.; et al. Effect of Early Particulate Air Pollution Exposure on Obesity in Mice. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 2518–2527.
- Mirmiran, P.; Hadavi, H.; Mottaghi, A.; Azizi, F. Advanced glycation end products and risk of general and abdominal obesity in Iranian adults: Tehran lipid and glucose study. Med. J. Islam. Repub. Iran 2019, 33, 21.
- Uribarri, J.; Woodruff, S.; Goodman, S.; Cai, W.; Chen, X.; Pyzik, R.; Yong, A.; Striker, G.E.; Vlassara, H. Advanced glycation end products in foods and a practical guide to their reduction in the diet. J. Am. Diet. Assoc. 2010, 110, 911–916.e12.
- Singh, R.; Barden, A.; Mori, T.; Beilin, L. Advanced glycation end-products: A review. Diabetologia 2001, 44, 129–146.
- Goldin, A.; Beckman, J.A.; Schmidt, A.M.; Creager, M.A. Advanced Glycation End Products. Circulation 2006, 114, 597–605.
- Aaseth, J.; Javorac, D.; Djordjevic, A.; Bulat, Z.; Skalny, A.; Zaitseva, I.; Aschner, M.; Tinkov, A. The Role of Persistent Organic Pollutants in Obesity: A Review of Laboratory and Epidemiological Studies. Toxics 2022, 10, 65.
- Hong, S.-H.; Sung, Y.-A.; Hong, Y.S.; Ha, E.; Jeong, K.; Chung, H.; Lee, H. Urinary bisphenol A is associated with insulin resistance and obesity in reproductive-aged women. Clin. Endocrinol. 2017, 86, 506–512.
- Wang, J.; Sun, B.; Hou, M.; Pan, X.; Li, X. The environmental obesogen bisphenol A promotes adipogenesis by increasing the amount of 11β-hydroxysteroid dehydrogenase type 1 in the adipose tissue of children. Int. J. Obes. 2013, 37, 999–1005.
- Amin, M.M.; Ebrahimpour, K.; Parastar, S.; Shoshtari-Yeganeh, B.; Hashemi, M.; Mansourian, M.; Poursafa, P.; Fallah, Z.; Rafiei, N.; Kelishadi, R. Association of urinary concentrations of phthalate metabolites with cardiometabolic risk factors and obesity in children and adolescents. Chemosphere 2018, 211, 547–556.
- Chiu, C.-Y.; Sun, S.-C.; Chiang, C.-K.; Wang, C.-C.; Chan, D.-C.; Chen, H.-J.; Liu, S.-H.; Yang, R.-S. Plasticizer di(2-ethylhexyl)phthalate interferes with osteoblastogenesis and adipogenesis in a mouse model. J. Orthop. Res. Off. Publ. Orthop. Res. Soc. 2018, 36, 1124–1134.
- Bimonte, V.M.; Besharat, Z.M.; Antonioni, A.; Cella, V.; Lenzi, A.; Ferretti, E.; Migliaccio, S. The endocrine disruptor cadmium: A new player in the pathophysiology of metabolic diseases. J. Endocrinol. Investig. 2021, 44, 1363–1377.
- Hurst, C.H.; Waxman, D.J. Activation of PPARalpha and PPARgamma by environmental phthalate monoesters. Toxicol. Sci. Off. J. Soc. Toxicol. 2003, 74, 297–308.
- Yang, C.; Kong, A.P.S.; Cai, Z.; Chung, A.C.K. Persistent Organic Pollutants as Risk Factors for Obesity and Diabetes. Curr. Diab. Rep. 2017, 17, 132.
- Henríquez-Hernández, L.A.; Luzardo, O.P.; Valerón, P.F.; Zumbado, M.; Serra-Majem, L.; Camacho, M.; González-Antuña, A.; Boada, L.D. Persistent organic pollutants and risk of diabetes and obesity on healthy adults: Results from a cross-sectional study in Spain. Sci. Total Environ. 2017, 607–608, 1096–1102.
- Pesta, M.; Cedikova, M.; Dvorak, P.; Dvorakova, J.; Kulda, V.; Srbecka, K.; Muller, L.; Bouchalova, V.; Kralickova, M.; Babuska, V.; et al. Trends in gene expression changes during adipogenesis in human adipose derived mesenchymal stem cells under dichlorodiphenyldichloroethylene exposure. Mol. Cell. Toxicol. 2018, 14, 369–379.
- Endocrine-Disrupting Chemicals—UpToDate. Available online: https://www.uptodate.com/contents/endocrine-disrupting-chemicals?search=EDCs%20and%20reproduction&source=search_result&selectedTitle=1~150&usage_type=default&display_rank=1 (accessed on 1 February 2023).
- Lim, J.-S.; Lee, D.-H.; Jacobs, D.R. Association of brominated flame retardants with diabetes and metabolic syndrome in the U.S. population, 2003–2004. Diabetes Care 2008, 31, 1802–1807.
- Helaleh, M.; Diboun, I.; Al-Tamimi, N.; Al-Sulaiti, H.; Al-Emadi, M.; Madani, A.; Mazloum, N.A.; Latiff, A.; Elrayess, M.A. Association of polybrominated diphenyl ethers in two fat compartments with increased risk of insulin resistance in obese individuals. Chemosphere 2018, 209, 268–276.
- Yang, C.; Wong, C.-M.; Wei, J.; Chung, A.C.K.; Cai, Z. The brominated flame retardant BDE 47 upregulates purine metabolism and mitochondrial respiration to promote adipocyte differentiation. Sci. Total Environ. 2018, 644, 1312–1322.
- Inoue, K.; Goto, A.; Sugiyama, T.; Ramlau-Hansen, C.H.; Liew, Z. The Confounder-Mediator Dilemma: Should We Control for Obesity to Estimate the Effect of Perfluoroalkyl Substances on Health Outcomes? Toxics 2020, 8, 125.
- Lee, D.-H.; Lee, I.-K.; Porta, M.; Steffes, M.; Jacobs, D.R. Relationship between serum concentrations of persistent organic pollutants and the prevalence of metabolic syndrome among non-diabetic adults: Results from the National Health and Nutrition Examination Survey 1999–2002. Diabetologia 2007, 50, 1841–1851.
- Scinicariello, F.; Buser, M.C. Urinary polycyclic aromatic hydrocarbons and childhood obesity: NHANES (2001–2006). Environ. Health Perspect. 2014, 122, 299–303.
- Fleisch, A.F.; Luttmann-Gibson, H.; Perng, W.; Rifas-Shiman, S.L.; Coull, B.A.; Kloog, I.; Koutrakis, P.; Schwartz, J.D.; Zanobetti, A.; Mantzoros, C.S.; et al. Prenatal and early life exposure to traffic pollution and cardiometabolic health in childhood. Pediatr. Obes. 2017, 12, 48–57.
- Fleisch, A.F.; Rifas-Shiman, S.L.; Koutrakis, P.; Schwartz, J.D.; Kloog, I.; Melly, S.; Coull, B.A.; Zanobetti, A.; Gillman, M.W.; Gold, D.R.; et al. Prenatal exposure to traffic pollution: Associations with reduced fetal growth and rapid infant weight gain. Epidemiol. Camb. Mass 2015, 26, 43–50.
- Fleisch, A.F.; Aris, I.M.; Rifas-Shiman, S.L.; Coull, B.A.; Luttmann-Gibson, H.; Koutrakis, P.; Schwartz, J.D.; Kloog, I.; Gold, D.R.; Oken, E. Prenatal Exposure to Traffic Pollution and Childhood Body Mass Index Trajectory. Front. Endocrinol. 2018, 9, 771.
- Wen, X.; Shenassa, E.D.; Paradis, A.D. Maternal smoking, breastfeeding, and risk of childhood overweight: Findings from a national cohort. Matern. Child Health J. 2013, 17, 746–755.
- Hawkins, S.S.; Cole, T.J.; Law, C. Millennium Cohort Study Child Health Group An ecological systems approach to examining risk factors for early childhood overweight: Findings from the UK Millennium Cohort Study. J. Epidemiol. Community Health 2009, 63, 147–155.
- Rundle, A.G.; Gallagher, D.; Herbstman, J.B.; Goldsmith, J.; Holmes, D.; Hassoun, A.; Oberfield, S.; Miller, R.L.; Andrews, H.; Widen, E.M.; et al. Prenatal exposure to airborne polycyclic aromatic hydrocarbons and childhood growth trajectories from age 5-14 years. Environ. Res. 2019, 177, 108595.
- Rundle, A.; Hoepner, L.; Hassoun, A.; Oberfield, S.; Freyer, G.; Holmes, D.; Reyes, M.; Quinn, J.; Camann, D.; Perera, F.; et al. Association of childhood obesity with maternal exposure to ambient air polycyclic aromatic hydrocarbons during pregnancy. Am. J. Epidemiol. 2012, 175, 1163–1172.
- Jerrett, M.; McConnell, R.; Wolch, J.; Chang, R.; Lam, C.; Dunton, G.; Gilliland, F.; Lurmann, F.; Islam, T.; Berhane, K. Traffic-related air pollution and obesity formation in children: A longitudinal, multilevel analysis. Environ. Health 2014, 13, 49.
- Lee, Y.-M.; Jacobs, D.R., Jr.; Lee, D.-H. Persistent Organic Pollutants and Type 2 Diabetes: A Critical Review of Review Articles. Front. Endocrinol. 2018, 9, 712.
- El-Sikaily, A.; Helal, M. Environmental pollution and diabetes mellitus. World J. Meta-Anal. 2021, 9, 220–326.
- Liu, S.; Guo, X.; Wu, B.; Yu, H.; Zhang, X.; Li, M. Arsenic induces diabetic effects through beta-cell dysfunction and increased gluconeogenesis in mice. Sci. Rep. 2014, 4, 6894.
- Rajak, S.; Raza, S.; Tewari, A.; Sinha, R.A. Environmental Toxicants and NAFLD: A Neglected yet Significant Relationship. Dig. Dis. Sci. 2022, 67, 3497–3507.
- Kalo, M.B.; Rezaei, M. In vitro toxic interaction of arsenic and hyperglycemia in mitochondria: An important implication of increased vulnerability in pre-diabetics. Environ. Sci. Pollut. Res. Vol. 2022, 29, 28375–28385.
- Hill, D.J. Impact of the exposome on the development and function of pancreatic β-cells. Mol. Aspects Med. 2022, 87, 100965.
- Khan, F.; Hodjat, M.; Rahimifard, M.; Nigjeh, M.N.; Azizi, M.; Baeeri, M.; Bayrami, Z.; Gholami, M.; Hassani, S.; Abdollahi, M. Assessment of arsenic-induced modifications in the DNA methylation of insulin-related genes in rat pancreatic islets. Ecotoxicol. Environ. Saf. 2020, 201, 110802.
- WHO. Preventing Disease Through Healthy Environments. Exposure to Cadmium: A Major Public Health Concern; WHO: Geneva, Switzerland, 2019; Available online: https://apps.who.int/iris/bitstream/handle/10665/329480/WHO-CED-PHE-EPE-19.4.3-eng.pdf (accessed on 20 September 2022).
- Xie, Y.; Liu, J.; Benbrahim-Tallaa, L.; Ward, J.M.; Logsdon, D.; Diwan, B.A.; Waalkes, M.P. Aberrant DNA methylation and gene expression in livers of newborn mice transplacentally exposed to a hepatocarcinogenic dose of inorganic arsenic. Toxicology 2007, 236, 7–15.
- Jacquet, A.; Arnaud, J.; Hininger-Favier, I.; Hazane-Puch, F.; Couturier, K.; Lénon, M.; Lamarche, F.; Ounnas, F.; Fontaine, E.; Moulis, J.-M.; et al. Impact of chronic and low cadmium exposure of rats: Sex specific disruption of glucose metabolism. Chemosphere 2018, 207, 764–773.
- Hong, H.; Xu, Y.; Xu, J.; Zhang, J.; Xi, Y.; Pi, H.; Yang, L.; Yu, Z.; Wu, Q.; Meng, Z.; et al. Cadmium exposure impairs pancreatic β-cell function and exaggerates diabetes by disrupting lipid metabolism. Environ. Int. 2021, 149, 106406.
- Buha, A.; Đukić-Ćosić, D.; Ćurčić, M.; Bulat, Z.; Antonijević, B.; Moulis, J.-M.; Goumenou, M.; Wallace, D. Emerging Links between Cadmium Exposure and Insulin Resistance: Human, Animal, and Cell Study Data. Toxics 2020, 8, 63.
- Little, B.B.; Reilly, R.; Walsh, B.; Vu, G.T. Cadmium Is Associated with Type 2 Diabetes in a Superfund Site Lead Smelter Community in Dallas, Texas. Int. J. Environ. Res. Public. Health 2020, 17, E4558.
- Han, J.C.; Park, S.Y.; Hah, B.G.; Choi, G.H.; Kim, Y.K.; Kwon, T.H.; Kim, E.K.; Lachaal, M.; Jung, C.Y.; Lee, W. Cadmium induces impaired glucose tolerance in rat by down-regulating GLUT4 expression in adipocytes. Arch. Biochem. Biophys. 2003, 413, 213–220.
- Chen, Y.W.; Yang, C.Y.; Huang, C.F.; Hung, D.Z.; Leung, Y.M.; Liu, S.H. Heavy metals, islet function and diabetes development. Islets 2009, 1, 169–176.
- Tinkov, A.A.; Filippini, T.; Ajsuvakova, O.P.; Aaseth, J.; Gluhcheva, Y.G.; Ivanova, J.M.; Bjørklund, G.; Skalnaya, M.G.; Gatiatulina, E.R.; Popova, E.V.; et al. The role of cadmium in obesity and diabetes. Sci. Total Environ. 2017, 601–602, 741–755.
- Nie, X.; Wang, N.; Chen, Y.; Chen, C.; Han, B.; Zhu, C.; Chen, Y.; Xia, F.; Cang, Z.; Lu, M.; et al. Blood cadmium in Chinese adults and its relationships with diabetes and obesity | SpringerLink. Available online: https://link.springer.com/article/10.1007/s11356-016-7078-2 (accessed on 20 September 2022).
- Zhang, S.; Jin, Y.; Zeng, Z.; Liu, Z.; Fu, Z. Subchronic Exposure of Mice to Cadmium Perturbs Their Hepatic Energy Metabolism and Gut Microbiome. Chem. Res. Toxicol. 2015, 28, 2000–2009.
- Treviño, S.; Waalkes, M.P.; Flores Hernández, J.A.; León-Chavez, B.A.; Aguilar-Alonso, P.; Brambila, E. Chronic cadmium exposure in rats produces pancreatic impairment and insulin resistance in multiple peripheral tissues. Arch. Biochem. Biophys. 2015, 583, 27–35.
- Sun, Q.; Yue, P.; Deiuliis, J.A.; Lumeng, C.N.; Kampfrath, T.; Mikolaj, M.B.; Cai, Y.; Ostrowski, M.C.; Lu, B.; Parthasarathy, S.; et al. Ambient Air Pollution Exaggerates Adipose Inflammation and Insulin Resistance in a Mouse Model of Diet-Induced Obesity. Circulation 2009, 119, 538–546.
- Acharjee, S.; Ghosh, B.; Al-Dhubiab, B.E.; Nair, A.B. Understanding Type 1 Diabetes: Etiology and Models. Can. J. Diabetes 2013, 37, 269–276.
- Hasham, A.; Tomer, Y. The recent rise in the frequency of Type 1 Diabetes: Who pulled the trigger? J. Autoimmun. 2011, 37, 1–2.
- Krämer, U.; Herder, C.; Sugiri, D.; Strassburger, K.; Schikowski, T.; Ranft, U.; Rathmann, W. Traffic-related air pollution and incident type 2 diabetes: Results from the SALIA cohort study. Environ. Health Perspect. 2010, 118, 1273–1279.
- Kelishadi, R.; Mirghaffari, N.; Poursafa, P.; Gidding, S.S. Lifestyle and environmental factors associated with inflammation, oxidative stress and insulin resistance in children. Atherosclerosis 2009, 203, 311–319.
- Novelli, M.; Beffy, P.; Masini, M.; Vantaggiato, C.; Martino, L.; Marselli, L.; Marchetti, P.; De Tata, V. Selective beta-cell toxicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin on isolated pancreatic islets. Chemosphere 2021, 265, 129103.
- Di Ciaula, A.; Portincasa, P. Relationships between emissions of toxic airborne molecules and type 1 diabetes incidence in children: An ecologic study. World J. Diabetes 2021, 12, 673–684.
- Hathout, E.H.; Beeson, W.L.; Nahab, F.; Rabadi, A.; Thomas, W.; Mace, J.W. Role of exposure to air pollutants in the development of type 1 diabetes before and after 5 yr of age. Pediatr. Diabetes 2002, 3, 184–188.
- Bresson, S.E.; Isom, S.; Jensen, E.T.; Huber, S.; Oulhote, Y.; Rigdon, J.; Lovato, J.; Liese, A.D.; Pihoker, C.; Dabelea, D.; et al. Associations between persistent organic pollutants and type 1 diabetes in youth. Environ. Int. 2022, 163, 107175.
- Fabricio, G.; Malta, A.; Chango, A.; De Freitas Mathias, P.C. Environmental Contaminants and Pancreatic Beta-Cells. J. Clin. Res. Pediatr. Endocrinol. 2016, 8, 257–263.
- Lin, Y.; Wei, J.; Li, Y.; Chen, J.; Zhou, Z.; Song, L.; Wei, Z.; Lv, Z.; Chen, X.; Xia, W.; et al. Developmental exposure to di(2-ethylhexyl) phthalate impairs endocrine pancreas and leads to long-term adverse effects on glucose homeostasis in the rat. Am. J. Physiol. Endocrinol. Metab. 2011, 301, E527–E538.
- Meier, J.J.; Köhler, C.U.; Alkhatib, B.; Sergi, C.; Junker, T.; Klein, H.H.; Schmidt, W.E.; Fritsch, H. Beta-cell development and turnover during prenatal life in humans. Eur. J. Endocrinol. 2010, 162, 559–568.
- Meier, J.J.; Butler, A.E.; Saisho, Y.; Monchamp, T.; Galasso, R.; Bhushan, A.; Rizza, R.A.; Butler, P.C. Beta-cell replication is the primary mechanism subserving the postnatal expansion of beta-cell mass in humans. Diabetes 2008, 57, 1584–1594.
- Yahaya, T.O.; Salisu, T.; Abdulrahman, Y.B.; Umar, A.K. Update on the genetic and epigenetic etiology of gestational diabetes mellitus: A review. Egypt. J. Med. Hum. Genet. 2020, 21, 13.
- Gestational Diabetes Mellitus (GDM). Available online: https://www.hopkinsmedicine.org/health/conditions-and-diseases/diabetes/gestational-diabetes (accessed on 25 August 2022).
- Butler, A.E.; Cao-Minh, L.; Galasso, R.; Rizza, R.A.; Corradin, A.; Cobelli, C.; Butler, P.C. Adaptive changes in pancreatic beta cell fractional area and beta cell turnover in human pregnancy. Diabetologia 2010, 53, 2167–2176.
- Robledo, C.A.; Mendola, P.; Yeung, E.; Männistö, T.; Sundaram, R.; Liu, D.; Ying, Q.; Sherman, S.; Grantz, K.L. Preconception and early pregnancy air pollution exposures and risk of gestational diabetes Mellitus. Environ. Res. 2015, 137, 316–322.
- Plows, J.F.; Stanley, J.L.; Baker, P.N.; Reynolds, C.M.; Vickers, M.H. The Pathophysiology of Gestational Diabetes Mellitus. Int. J. Mol. Sci. 2018, 19, 3342.
- Zhang, M.; Wang, X.; Yang, X.; Dong, T.; Hu, W.; Guan, Q.; Tun, H.M.; Chen, Y.; Chen, R.; Sun, Z.; et al. Increased risk of gestational diabetes mellitus in women with higher prepregnancy ambient PM2.5 exposure. Sci. Total Environ. 2020, 730, 138982.
- Ramanjaneya, M.; Butler, A.E.; Alkasem, M.; Bashir, M.; Jerobin, J.; Godwin, A.; Moin, A.S.M.; Ahmed, L.; Elrayess, M.A.; Hunt, S.C.; et al. Association of Complement-Related Proteins in Subjects With and Without Second Trimester Gestational Diabetes. Front. Endocrinol. 2021, 12, 641361. Available online: https://www.frontiersin.org/articles/10.3389/fendo.2021.641361 (accessed on 29 September 2022).
- Ramanjaneya, M.; Butler, A.E.; Bashir, M.; Bettahi, I.; Moin, A.S.M.; Ahmed, L.; Elrayess, M.A.; Hunt, S.C.; Atkin, S.L.; Abou-Samra, A.B. apoA2 correlates to gestational age with decreased apolipoproteins A2, C1, C3 and E in gestational diabetes. BMJ Open Diabetes Res. Care 2021, 9, e001925.
- Zhang, H.; Wang, Q.; He, S.; Wu, K.; Ren, M.; Dong, H.; Di, J.; Yu, Z.; Huang, C. Ambient air pollution and gestational diabetes mellitus: A review of evidence from biological mechanisms to population epidemiology. Sci. Total Environ. 2020, 719, 137349.
- Lawrence, R.L.; Wall, C.R.; Bloomfield, F.H. Prevalence of gestational diabetes according to commonly used data sources: An observational study. BMC Pregnancy Childbirth 2019, 19, 349.
- Zhou, T.; Sun, D.; Li, X.; Heianza, Y.; Nisa, H.; Hu, G.; Pei, X.; Shang, X.; Qi, L. Prevalence and Trends in Gestational Diabetes Mellitus among Women in the United States, 2006–2016. Diabetes 2018, 67, 121-OR.
- Gao, C.; Sun, X.; Lu, L.; Liu, F.; Yuan, J. Prevalence of gestational diabetes mellitus in mainland China: A systematic review and meta-analysis. J. Diabetes Investig. 2019, 10, 154–162.
- Al-Rifai, R.H.; Abdo, N.M.; Paulo, M.S.; Saha, S.; Ahmed, L.A. Prevalence of Gestational Diabetes Mellitus in the Middle East and North Africa, 2000–2019: A Systematic Review, Meta-Analysis, and Meta-Regression. Front. Endocrinol. 2021, 12, 668447. Available online: https://www.frontiersin.org/articles/10.3389/fendo.2021.668447 (accessed on 16 September 2022).
- Paulo, M.S.; Abdo, N.M.; Bettencourt-Silva, R.; Al-Rifai, R.H. Gestational Diabetes Mellitus in Europe: A Systematic Review and Meta-Analysis of Prevalence Studies. Front. Endocrinol. 2021, 12, 691033.
- Zhu, Y.; Zhang, C. Prevalence of Gestational Diabetes and Risk of Progression to Type 2 Diabetes: A Global Perspective. Curr. Diab. Rep. 2016, 16, 7.
- Lavigne, E.; Ashley-Martin, J.; Dodds, L.; Arbuckle, T.E.; Hystad, P.; Johnson, M.; Crouse, D.L.; Ettinger, A.S.; Shapiro, G.D.; Fisher, M.; et al. Air Pollution Exposure During Pregnancy and Fetal Markers of Metabolic function. Am. J. Epidemiol. 2016, 183, 842–851.
- Shapiro, G.D.; Dodds, L.; Arbuckle, T.E.; Ashley-Martin, J.; Ettinger, A.S.; Fisher, M.; Taback, S.; Bouchard, M.F.; Monnier, P.; Dallaire, R.; et al. Exposure to organophosphorus and organochlorine pesticides, perfluoroalkyl substances, and polychlorinated biphenyls in pregnancy and the association with impaired glucose tolerance and gestational diabetes mellitus: The MIREC Study. Environ. Res. 2016, 147, 71–81.
- Eslami, B.; Naddafi, K.; Rastkari, N.; Rashidi, B.H.; Djazayeri, A.; Malekafzali, H. Association between serum concentrations of persistent organic pollutants and gestational diabetes mellitus in primiparous women. Environ. Res. 2016, 151, 706–712.
- Vafeiadi, M.; Roumeliotaki, T.; Chalkiadaki, G.; Rantakokko, P.; Kiviranta, H.; Fthenou, E.; Kyrtopoulos, S.A.; Kogevinas, M.; Chatzi, L. Persistent organic pollutants in early pregnancy and risk of gestational diabetes mellitus. Environ. Int. 2017, 98, 89–95.
- Rahman, M.L.; Zhang, C.; Smarr, M.M.; Lee, S.; Honda, M.; Kannan, K.; Tekola-Ayele, F.; Buck Louis, G.M. Persistent organic pollutants and gestational diabetes: A multi-center prospective cohort study of healthy US women. Environ. Int. 2019, 124, 249–258.