2. cGAS-STING Canonical Signaling Pathway
Among all cellular DNA sensors, cGAS is the best characterized and considered essential for developing an innate immune response against cytosolic dsDNA [
25,
26]. This protein can detect pathogenic DNA or DNA released from the mitochondria or the nucleus after cell damage, even when it has been oxidized [
25,
27]. dsDNA fragments of more than 20 base pairs (bp) can be recognized by cGAS in a sequence-independent manner inducing its dimerization in a 2:2 DNA/cGAS complex [
25,
28]. Fragments of DNA smaller than 20 bp are recognized by cGAS but are unable to induce dimerization and thus do not induce its activation, while long chains of dsDNA induce the formation of ladder-like structures that result in a stronger activation of this sensor [
25,
29].
After the formation of dimers or higher structures, cGAS changes conformation enabling its catalytic site activation (
Figure 1) [
25,
28]. Once activated, cGAS can use adenine triphosphate (ATP) and guanine triphosphate (GTP) as substrates to produce the secondary metabolite cyclic cGAMP [
25,
26,
28]. cGAMP produced by cGAS is a CDN that contains two phosphodiester bonds, one between the 2′-OH of GMP and the 5′-phosphate of AMP and the other between the 3′-OH of AMP and the 5′-phosphate of GMP (2′3′-cGAMP). This ring-structured molecule acts as a second messenger, binding and inducing STING activation. It has also been described that cGAMP can pass to neighboring cells through gap junctions in a process dependent on connexin 43 allowing contacting cells to trigger STING activation [
30].
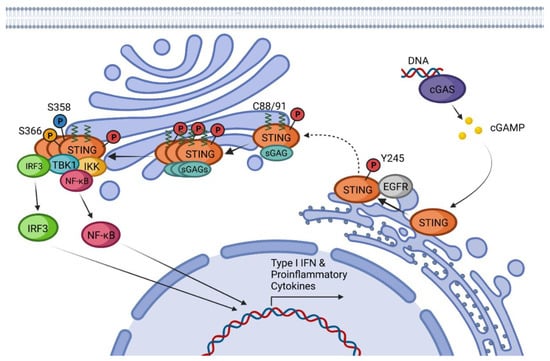
Figure 1. cGAS-STING canonical pathway: Schematic representation of STING canonical activation including post-translational modifications, modulator proteins, and main effectors involved in the production of Type I IFN and proinflammatory cytokines.
When first described, STING was proposed to directly recognize bacteria-derived CDNs such as cdiGMP, cdiAMP, and 3′3′-cGAMP, which can act as PAMPs to activate the immune responses during infection [
31].
In addition to interacting with STING in host cells, CDNs play a pivotal role as second messengers controlling physiological processes in bacteria [
32,
33]. Moreover, these molecules appear to modulate many behaviors facets at the bacteria community level, such as
quorum sensing, the formation of aggregates, swarming motility, or the formation of bacterial biofilms [
34,
35]. There is a correlation between high intracellular levels of cdiGMP and biofilm formation and a sessile lifestyle. In contrast, low cdiGMP levels are associated with a motile or planktonic existence [
36]. 3′3′-cGAMP also has been related to protection against phage infections in bacteria [
37].
Inactive resting STING forms a homodimer that suffers a 180° twist and several conformational changes upon CDN-binding [
38,
39]. Conformational changes of STING after CDN activation promote the binding to the epidermal growth factor receptor (EGFR) in the ER, causing EGFR auto-phosphorylation at Y1068 and its consequent activation. Activated EGFR also phosphorylates STING at Y245 [
40]. STING translocates from the ER to the Golgi apparatus by passing through the ER-Golgi intermediate compartment (ERGIC) [
41]. The translocation process is driven by the STING-iRhom2-TRAPβ complex [
42]. It is also known that K63-linked ubiquitination is required for STING trafficking, but it is not completely clear if this modification is produced on resting STING or after activation by CDNs [
43].
Once in the Golgi, STING binds to sulfated glycosaminoglycans (sGAGs) and is palmitoylated in C88 and C91, with these two modifications being necessary to induce STING polymerization and the formation of oligomers [
44,
45]. Mukai et al. demonstrated by tritylation experiments that of the 23 human-encoded palmitoyl transferases (DHHCs), those responsible for the palmitoylation of STING are DHHC3, DHHC7, and DHHC15 [
45]. This oligomerization is required for the signal transduction and activation of STING effectors [
44,
45]. It has been proposed that C88 and C91 palmitoylation induces the STING clustering into lipid rafts from the cytosolic side of the Golgi while sGAGs promote STING polymerization from the lumen [
45]. After oligomerization, STING recruits and activates the downstream effector kinase TBK1 [
28]. Activated TBK1 dimerizes and induces its autophosphorylation and phosphorylation of STING in two different residues, S358 and S366 [
28,
46,
47,
48]. As the first step, TBK1 phosphorylates S358, stabilizing the STING-TBK1 complex. The kinase domain of TBK1 cannot phosphorylate S366 since it is bound to this region of STING. To achieve S366 phosphorylation, TBK1 requires the previous formation of STING oligomers so that the kinase domain reaches S366 of the neighboring STING-TBK1 complexes [
46].
The TBK1-STING phosphorylated complex recruits and phosphorylates Interferon Regulatory Factor 3 (IRF3) inducing its dimerization and translocation into the nucleus, which results in the expression of type I IFNs, a set of Interferon-Stimulated Genes (ISGs) and proinflammatory cytokines [
49]. IRF3 is not the only transcription factor that STING can recruit. STING activation has been shown to induce the activation of IKKε, which in a redundant combination with TBK1 can induce the phosphorylation and translocation of NF-κB [
50]. STING-dependent activation of NF-κB is responsible for the production of proinflammatory cytokines such as IL-6, IL-12, or TNF-α [
28,
51]. All these effects together lead to the development of an antiviral state and trigger innate and adaptive immune responses.
STING activation has been linked to other biological effects in addition to IFN and proinflammatory cytokines production. It is well known that STING activation triggers autophagy in a process that is independent of TBK1, IFN, and the Unc-51-Like Autophagy Activating Kinase (ULK) [
28,
52,
53]. STING induction of autophagy is necessary for cytosolic DNA and virus clearance and the depletion of activated STING structures [
30,
52]. Once STING is activated, it initiates a translocation from ER to the Golgi apparatus as described before. During this process, STING passes through the ERGIC intermediate compartment in a coat complex protein II (COPII) and ADP-ribosylation factor (ARF) GTPase-dependent manner [
52]. STING containing ERGIC induces light chain 3 (LC3) lipidation into LC3-II, a classic effector of autophagy, in a process dependent on ATG5 and WIPI2 [
30,
52,
54]. Although autophagy has not been related to the induction of type I IFN expression, its implication in the IFN system seems to provide a downregulation of PRRs and receptors whose abundance is necessary to avoid tissue damage produced by excessive immune stimulation [
55].
3. STING Non-Canonical Signaling Pathway
The activation of STING is possible even in the absence of cGAS, 2′3′-cGAMP, or other CDNs as inducers (
Figure 2) [
56,
57]. Until now, different cGAS-independent mechanisms of STING activation have been described.
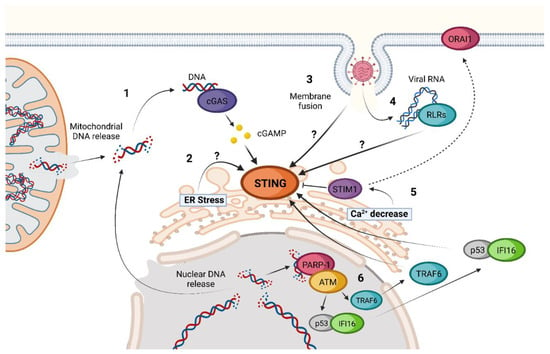
Figure 2. Non-canonical activations of STING. 1. Release of nuclear or mitochondrial DNA into the cytoplasm during viral infection or after cell damage. 2. Activation of the STING–TBK1–IRF3 axis by ER-stress inducers such as ethanol. 3. cGAS-independent activation of STING triggered by membrane fusion. 4. Activation of STING by RIG-I-like Receptors (RLRs) after detection of viral RNA in a MAVS-independent process. 5. Ca2+ depletion in the ER triggers activation of STIM1 sensor, an inhibitor of STING, and induces its migration to join the calcium channel ORAI1, increasing STING activity. 6. IFI16 pathway of nuclear-damaged DNA detection in the absence of cGAS activation. Question marks (?) represent “unknown activation mechanism”.
Interferon Gamma Inducible Protein 16 (IFI16) is a DNA sensor located in the cytosol and the nucleus and has been proven to be both a coactivator in the classic cGAS-STING pathway and a cGAS-independent STING inducer [
58]. In the canonical pathway, IFI16 is necessary for the 2′3′-cGAMP production by stabilizing the cGAS–DNA interaction and for the STING activation of TBK1, at least in some cell types [
58,
59].
The proposed IFI16-dependent non-canonical activation of STING begins with polyADP-ribose polymerase-1 (PARP-1) and ataxia telangiectasia mutated (ATM) detection of damaged DNA in the nucleus. After this recognition, ATM phosphorylates p53, inducing the recruitment of IFI16. The complex p53-IFI16 translocases to the cytoplasm and is capable of interacting with TRAF6. Once activated, TRAF6 induces the formation of multiple K63-linked ubiquitin chains on STING. This ubiquitination leads to a non-canonical process of STING activation [
58]. IFI16-dependent activation of STING takes place without STING phosphorylation in S366, TBK1 recruitment, or trafficking to ERGIC, leading to a predominant activation of NF-κB instead of IRF3 as compared to the canonical cGAS-STING pathway [
58].
Another non-canonical activation of STING that has been proposed is through ER stress induction [
57]. It has been suggested that ethanol can induce STING-TBK1-IRF3 activation in a not-very-well-characterized ER stress-dependent activation. This activation leads to IRF3-dependent Type I IFN induction and apoptosis through B-cell lymphoma 2 (Bcl2)-associated X protein (BAX) activation [
57]. Many viruses, such as herpes simplex virus (HSV) or West Nile virus (WNV), amongst others [
60], can induce ER stress during infection [
61,
62]. Thus, understanding how this ER stress-dependent activation of STING works can be important to understand better host defense against infections.
Calcium cell homeostasis seems to influence STING activation as well [
57]. Stromal Interaction Molecule 1 (STIM1) is a Ca
2+ sensor that is in the ER and interacts with inactivated STING. Due to this interaction, inactivated STIM1 acts as a natural inhibitor of STING oligomerization and trafficking [
56,
57]. STIM1 knock-out cells have basal levels of STING activation and IFN expression that are higher than those present in cells that present a normal STIM1 expression [
56,
57]. Under ER Ca
2+ depletion conditions, STIM1 suffers a conformational change that enables the interaction with calcium release-activated calcium channel protein 1 (ORAI1) [
63]. This way, STIM1 has been proposed separately to facilitate STING activation. As for the case of ER stress, many viruses have shown ER Ca
2+ alterations during infection, making it another possibility for triggering innate immunity through these changes [
64].
Different studies have linked STING activation to RNA virus infections [
62,
65,
66,
67]. A hint of the relevance of STING in the innate immune response to RNA virus infection is that many RNA viruses have developed mechanisms to inhibit STING pathway activation [
68,
69,
70,
71].
Mechanisms of STING activation by these viruses seem diverse, and some remain unclear. One of the most evident is the indirect activation of the classical cGAS-STING pathway by the detection of mitochondrial DNA (mDNA) released due to the increase in ROS and the activation of the inflammasome during dengue virus (DENV) infection [
72,
73]. Another example is the detection of damaged nuclear DNA as a result of SARS-CoV-2 spike protein expression associated with infection. Due to its main role as a fusion protein, the expression of the Spike under SARS-CoV-2 infection leads to the formation of multinucleated syncytial cells in which damaged nuclei produce micronuclei, which, in turn, are detected by cGAS [
74,
75]. Meanwhile, membrane fusion has been proven to induce cGAS-independent STING activation that leads to TBK1 and IRF3 activation and the type I IFN response [
76,
77]. Influenza A virus (IAV) blocks this signaling through a direct interaction between Hemagglutinin (HA) fusion peptide (FP) and STING, preventing TBK1 activation and STING phosphorylation [
77]. The exact mechanism by which membrane fusion activates STING has not been addressed. Another role of STING under RNA viral infections seems to be blocking the translation of viral and host proteins [
67]. This activation of STING is dependent on classic cytosolic RNA sensors and RLR but is MAVS-independent and does not result in IFN expression or autophagy induction [
67]. Different studies have shown this link between STING and RLR, but the mechanism is still unknown [
1,
78].