Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Irinotecan (SN-38) is a potent and broad-spectrum anticancer drug that targets DNA topoisomerase I (Top1). It exerts its cytotoxic effects by binding to the Top1-DNA complex and preventing the re-ligation of the DNA strand, leading to the formation of lethal DNA breaks. Following the initial response to irinotecan, secondary resistance is acquired relatively rapidly, compromising its efficacy. There are several mechanisms contributing to the resistance, which affect the irinotecan metabolism or the target protein.
- irinotecan
- drug resistance
- topoisomerase I
- dose escalation
1. Introduction
Irinotecan (CPT-11) is a chemotherapeutic agent that causes cancer cell killing by poisoning topoisomerase I (Top1) in the cell. It is a semisynthetic analog of camptothecin, which was originally isolated from the Chinese/Tibetan ornamental tree Camptotheca acuminata [1][2][3]. Resistance model-based studies uncovered several mechanisms of cellular resistance to this agent and, accordingly, multiple approaches were tested in clinical trials to circumvent the resistance [4][5][6]. Beside primary resistance, the major clinical problem is development of secondary resistance in the course of the drug treatment, which is often observed with DNA-damaging chemotherapeutic drugs, such as irinotecan [7][8][9] or doxorubicin [6][10][11]. It is commonly understood that the development of drug resistance in cancer cells is defined by the change in expression or function of the target protein [8][12], changes in the ability of cells to undergo cell death [13][14][15], or changes in drug metabolism [16][17][18][19].
Importantly, both in experimental and clinical settings, secondary resistance usually develops gradually in the course of multiple exposures to the drug. It is unclear why changes in transcriptome [9][12][16][18][19][20][21], metabolism [12][22][23], or mutations in drug targets [7][12][24][25][26][27][28][29][30] do not appear abruptly but require multiple drug administrations [12][14][16][31][32]. This puzzling requirement suggests a distinct mode of resistance that may require a gradual accumulation of a large number of mutations or epigenetic events. Each of these events may have a minor effect on the drug response, but cumulatively they provide significant resistance.
2. Irinotecan’s Mode of Action
One of the classes of drugs that are frequently used in cancer therapy is inhibitors of DNA supercoil relaxing topoisomerases, including Top1 and Top2. Type I topoisomerases (Top1) cause relaxation of super helical DNA by generating a transient single-strand nick, followed by DNA relaxation and re-ligation. The enzyme subtypes perform very specialized functions, e.g., Class IA can only relax negative supercoiled DNA, whereas Class IB can introduce positive supercoils, relaxing and separating DNA molecules in daughter chromosomes after DNA replication. Top1 plays major role in transcription and replication and is highly active in pericentromeric and centromeric regions of the genome [33][34][35][36][37][38][39]. In contrast, type II topoisomerases (Top2) mediate ATP-dependent cleavage of both strands of the DNA double helix, followed by crossing of the DNA double-strand (ds) through the transiently opened gap [40]. Mammalian cells have two Top2 isoenzymes, Top2α and Top2β [35]. Top2α is associated with DNA replication and, as a consequence, cell proliferation, while Top2β may play a role in transcription [39][41].
Major Top1 or Top2 inhibitors function by stabilizing the transient complexes formed between these enzymes and DNA [37][42]. Stabilization of these otherwise fleeting “cleavable complexes” can lead to formation of double-strand breaks (DSBs) when the DNA replication forks collide with the topoisomerase-DNA complexes [37][42]. Similarly, the inhibitor-stabilized Top1 or Top2 “cleavable complexes” located on the transcribed template strand can also result in the formation of DSBs when RNA polymerase molecules collide with them within the transcribed DNA regions [40]. DSBs are recognized by the cell as lethal lesions and can trigger apoptosis [43][44]. These cytotoxic effects are responsible for the anti-cancer activity of topoisomerase inhibitors.
A most widely used Top1 inhibitor, irinotecan is a prodrug that, upon activation, generates an active compound, SN-38 [2][45][46][47][48]. Upon treatment of cancer cells, SN-38 binds to Top1 and stabilizes Top1-DNA complexes [2][49] (Figure 1). The SN-38 molecule stacks against the base pairs flanking the Top1-induced cleavage site and poisons the enzyme [2][49]. This conversion suppresses the 3′-OH free end and makes it unavailable for re-ligation. Inhibition of re-ligation of nicked DNA strands leads to single strand breaks, which have a high probability of conversion to highly toxic DSB. As the drug works not only during transcription [50], but also during replication, these DNA-Top1-SN38 covalent adjuncts could lead to replication fork stalling and the arrest of DNA replication.
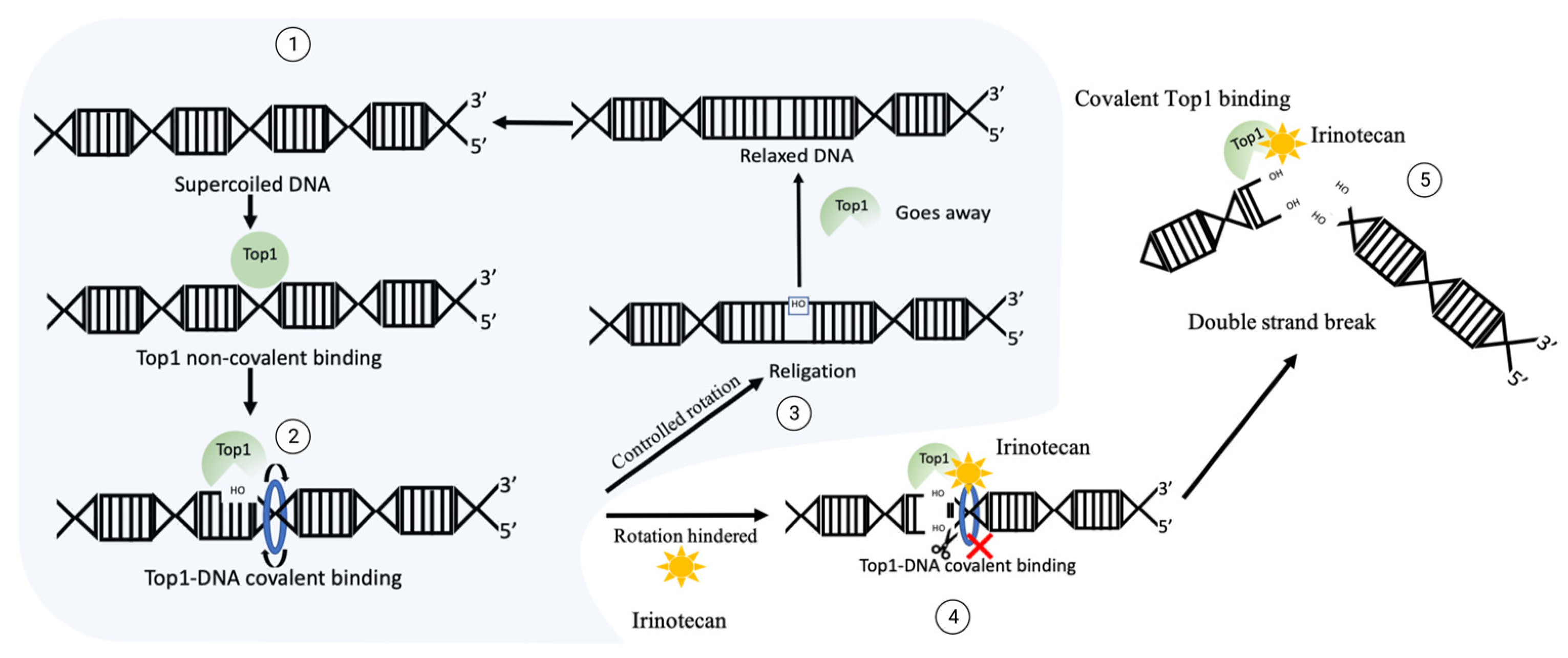
Figure 1. The mechanism of DNA generation breaks upon irinotecan exposure. Normal function of Top1: ① supercoiled sensing and nick development by Top1 leads to ② relaxation, then ③ Top1 re-ligates the nicked backbone and leaves the site. In the case of irinotecan exposure, Top1 gets covalently crosslinked to DNA, then ④ ligation of 3′-OH free end is blocked, which could translate into ⑤ double strand break and facilitate cytotoxicity.
High-intensity transcription and replication enhances the supercoiling of DNA to levels that can impede or halt these processes. As a potent transcription amplifier and replication accelerator, the proto-oncogene MYC must manage this interfering torsional change. In a recent study, a direct association of MYC with Top1 and Top2 was demonstrated [51]. Beyond recruiting topoisomerases, MYC directly stimulated their activities. These MYC complexes with Top1 and Top2 increased their activities at promoters, coding regions, and enhancers [51]. Such enhancement of the activity of Top1 and its recruitment to DNA may create additional cleavage sites upon the drug treatment. In line with this suggestion, it was demonstrated that the overexpression of MYC enhances the sensitivity of colon cancer cells to the parental drug, camptothecin [51][52]. At the same time, MYC can activate the DNA damage response, which results in induction of the DSB repair system [51]. This in turn could reduce the response to irinotecan, since DSB repair plays a critical role in survival of cells treated with irinotecan.
3. Clinical Use of Irinotecan
In the USA, irinotecan has been approved for use against colorectal cancer in combination with 5-fluorouracil (5-FU) and leucovorin (FOLFIRI regimen). With therapy regimens like FOLFIRI, the median survival rate of a patient with metastatic colorectal cancer has improved from 8 months to 24 months [47]. Irinotecan is also used in combination with Capecitabine (pro-drug of 5-FU) (XELIRI regimen). Currently, both regimens are considered first-line therapy for cancer treatment. Several studies have been conducted to assess the effects of these combinations, and they have demonstrated that they are equally effective, with certain variations in median survival rates [1][53][54][55][56][57][58][59]. The usage of these schemes is defined by several factors such as geographical regions, patient genetics, individual response rate, oncologist’s preference, and socio-economic factors.
Another promising combination of irinotecan is with antibodies against EGFR, such as Cetuximab, for treatment of patients with wild-type K-Ras colorectal cancers and certain other cancer types, with 5-FU/leucovorin as a first-line treatment [60]. It is important to note that initial studies suggested that patients with colorectal tumors characterized by high microsatellite instability (MSI) might respond better to irinotecan-based chemotherapy [61][62]. However, subsequent data did not support a predictive value of MSI status in relation to treatment response [63][64].
4. Resistance development
There are several standard mechanisms of resistance to the drug. Among which MDRs or mutations in the target are prevalent. In addition, there is a unique mechanism of rapid development of resistance associated with the accumulation of multiple mutations resulting from DSB repair of Top1-generated DNA breaks[65]. These mutations prevent interaction of Top1 with these specific sites upon following exposure to irinotecan and thus lead to resistance.
This entry is adapted from the peer-reviewed paper 10.3390/ijms24087233
References
- Lavelle, F.; Bissery, M.C.; André, S.; Roquet, F.; Riou, J.F. Preclinical Evaluation of CPT-11 and Its Active Metabolite SN-38. Semin. Oncol. 1996, 23, 11–20.
- Xu, Y.; Villalona-Calero, M.A. Irinotecan: Mechanisms of Tumor Resistance and Novel Strategies for Modulating Its Activity. Ann. Oncol. 2002, 13, 1841–1851.
- Bailly, C. Irinotecan: 25 Years of Cancer Treatment. Pharmacol. Res. 2019, 148, 104398.
- Ahronian, L.G.; Corcoran, R.B. Strategies for Monitoring and Combating Resistance to Combination Kinase Inhibitors for Cancer Therapy. Genome Med. 2017, 9, 37.
- Wang, X.; Zhang, H.; Chen, X. Drug Resistance and Combating Drug Resistance in Cancer. Cancer Drug Resist. 2019, 2, 141–160.
- Dragojevic, S.; Turner, L.; Raucher, D. Circumventing Doxorubicin Resistance Using Elastin-like Polypeptide Biopolymer-Mediated Drug Delivery. Int. J. Mol. Sci. 2022, 23, 2301.
- Arakawa, Y.; Suzuki, H.; Saito, S.; Yamada, H. Novel Missense Mutation of the DNA Topoisomerase I Gene in SN-38-Resistant DLD-1 Cells. Mol. Cancer Ther. 2006, 5, 502–508.
- Sugimoto, Y.; Tsukahara, S.; Oh-Hara, T.; Isoe, T.; Tsuruo, T. Decreased Expression of DNA Topoisomerase I in Camptothecin-Resistant Tumor Cell Lines as Determined by a Monoclonal Antibody. Cancer Res. 1990, 50, 6925–6930.
- Cummings, J.; Boyd, G.; Ethell, B.T.; Macpherson, J.S.; Burchell, B.; Smyth, J.F.; Jodrell, D.I. Enhanced Clearance of Topoisomerase I Inhibitors from Human Colon Cancer Cells by Glucuronidation. Biochem. Pharmacol. 2002, 63, 607–613.
- Tacar, O.; Sriamornsak, P.; Dass, C.R. Doxorubicin: An Update on Anticancer Molecular Action, Toxicity and Novel Drug Delivery Systems. J. Pharm. Pharmacol. 2013, 65, 157–170.
- Pang, B.; Qiao, X.; Janssen, L.; Velds, A.; Groothuis, T.; Kerkhoven, R.; Nieuwland, M.; Ovaa, H.; Rottenberg, S.; van Tellingen, O.; et al. Drug-Induced Histone Eviction from Open Chromatin Contributes to the Chemotherapeutic Effects of Doxorubicin. Nat. Commun. 2013, 4, 1908.
- Jensen, N.F.; Agama, K.; Roy, A.; Smith, D.H.; Pfister, T.D.; Rømer, M.U.; Zhang, H.-L.; Doroshow, J.H.; Knudsen, B.R.; Stenvang, J.; et al. Characterization of DNA Topoisomerase I in Three SN-38 Resistant Human Colon Cancer Cell Lines Reveals a New Pair of Resistance-Associated Mutations. J. Exp. Clin. Cancer Res. 2016, 35, 56.
- Koizumi, N.; Hatano, E.; Nitta, T.; Tada, M.; Harada, N.; Taura, K.; Ikai, I.; Shimahara, Y. Blocking of PI3K/Akt Pathway Enhances Apoptosis Induced by SN-38, an Active Form of CPT-11, in Human Hepatoma Cells. Int. J. Oncol. 2005, 26, 1301–1306.
- Tamura, N.; Hirano, K.; Kishino, K.; Hashimoto, K.; Amano, O.; Shimada, J.; Sakagami, H. Analysis of Type of Cell Death Induced by Topoisomerase Inhibitor SN-38 in Human Oral Squamous Cell Carcinoma Cell Lines. Anticancer Res. 2012, 32, 4823–4832.
- Ueno, M.; Nonaka, S.; Yamazaki, R.; Deguchi, N.; Murai, M. SN-38 Induces Cell Cycle Arrest and Apoptosis in Human Testicular Cancer. Eur. Urol. 2002, 42, 390–397.
- Innocenti, F.; Schilsky, R.L.; Ramírez, J.; Janisch, L.; Undevia, S.; House, L.K.; Das, S.; Wu, K.; Turcich, M.; Marsh, R.; et al. Dose-Finding and Pharmacokinetic Study to Optimize the Dosing of Irinotecan According to the UGT1A1 Genotype of Patients with Cancer. J. Clin. Oncol. 2014, 32, 2328–2334.
- Oguri, T.; Takahashi, T.; Miyazaki, M.; Isobe, T.; Kohno, N.; Mackenzie, P.I.; Fujiwara, Y. UGT1A10 Is Responsible for SN-38 Glucuronidation and Its Expression in Human Lung Cancers. Anticancer Res. 2004, 24, 2893–2896.
- Olszewski, U.; Liedauer, R.; Ausch, C.; Thalhammer, T.; Hamilton, G. Overexpression of CYP3A4 in a COLO 205 Colon Cancer Stem Cell Model in Vitro. Cancers 2011, 3, 1467–1479.
- Van der Bol, J.M.; Mathijssen, R.H.J.; Creemers, G.-J.M.; Planting, A.S.T.; Loos, W.J.; Wiemer, E.A.C.; Friberg, L.E.; Verweij, J.; Sparreboom, A.; de Jong, F.A. A CYP3A4 Phenotype-Based Dosing Algorithm for Individualized Treatment of Irinotecan. Clin. Cancer Res. 2010, 16, 736–742.
- Ohtsuka, K.; Inoue, S.; Kameyama, M.; Kanetoshi, A.; Fujimoto, T.; Takaoka, K.; Araya, Y.; Shida, A. Intracellular Conversion of Irinotecan to Its Active Form, SN-38, by Native Carboxylesterase in Human Non-Small Cell Lung Cancer. Lung Cancer 2003, 41, 187–198.
- Robey, R.W.; Medina-Pérez, W.Y.; Nishiyama, K.; Lahusen, T.; Miyake, K.; Litman, T.; Senderowicz, A.M.; Ross, D.D.; Bates, S.E. Overexpression of the ATP-Binding Cassette Half-Transporter, ABCG2 (Mxr/BCrp/ABCP1), in Flavopiridol-Resistant Human Breast Cancer Cells. Clin. Cancer Res. 2001, 7, 145–152.
- Mittal, B.; Tulsyan, S.; Kumar, S.; Mittal, R.D.; Agarwal, G. Cytochrome P450 in Cancer Susceptibility and Treatment. Adv. Clin. Chem. 2015, 71, 77–139.
- Dean, L. Irinotecan Therapy and UGT1A1 Genotype. In Medical Genetics Summaries; Pratt, V.M., Scott, S.A., Pirmohamed, M., Esquivel, B., Kattman, B.L., Malheiro, A.J., Eds.; National Center for Biotechnology Information: Bethesda, MD, USA, 2012.
- Gongora, C.; Vezzio-Vie, N.; Tuduri, S.; Denis, V.; Causse, A.; Auzanneau, C.; Collod-Beroud, G.; Coquelle, A.; Pasero, P.; Pourquier, P.; et al. New Topoisomerase I Mutations Are Associated with Resistance to Camptothecin. Mol. Cancer 2011, 10, 64.
- Okubo, M.; Murayama, N.; Shimizu, M.; Shimada, T.; Guengerich, F.P.; Yamazaki, H. The CYP3A4 Intron 6 C>T Polymorphism (CYP3A4*22) Is Associated with Reduced CYP3A4 Protein Level and Function in Human Liver Microsomes. J. Toxicol. Sci. 2013, 38, 349–354.
- Maekawa, K.; Harakawa, N.; Yoshimura, T.; Kim, S.-R.; Fujimura, Y.; Aohara, F.; Sai, K.; Katori, N.; Tohkin, M.; Naito, M.; et al. CYP3A4*16 and CYP3A4*18 Alleles Found in East Asians Exhibit Differential Catalytic Activities for Seven CYP3A4 Substrate Drugs. Drug Metab. Dispos. 2010, 38, 2100–2104.
- Lévesque, É.; Bélanger, A.-S.; Harvey, M.; Couture, F.; Jonker, D.; Innocenti, F.; Cecchin, E.; Toffoli, G.; Guillemette, C. Refining the UGT1A Haplotype Associated with Irinotecan-Induced Hematological Toxicity in Metastatic Colorectal Cancer Patients Treated with 5-Fluorouracil/Irinotecan-Based Regimens. J. Pharmacol. Exp. Ther. 2013, 345, 95–101.
- Gagné, J.-F.; Montminy, V.; Belanger, P.; Journault, K.; Gaucher, G.; Guillemette, C. Common Human UGT1A Polymorphisms and the Altered Metabolism of Irinotecan Active Metabolite 7-Ethyl-10-Hydroxycamptothecin (SN-38). Mol. Pharmacol. 2002, 62, 608–617.
- Desai, A.A.; Innocenti, F.; Ratain, M.J. UGT Pharmacogenomics: Implications for Cancer Risk and Cancer Therapeutics. Pharmacogenetics 2003, 13, 517–523.
- Kim, S.-R.; Sai, K.; Tanaka-Kagawa, T.; Jinno, H.; Ozawa, S.; Kaniwa, N.; Saito, Y.; Akasawa, A.; Matsumoto, K.; Saito, H.; et al. Haplotypes and a Novel Defective Allele of CES2 Found in a Japanese Population. Drug Metab. Dispos. 2007, 35, 1865–1872.
- Vitiello, P.P.; Martini, G.; Mele, L.; Giunta, E.F.; De Falco, V.; Ciardiello, D.; Belli, V.; Cardone, C.; Matrone, N.; Poliero, L.; et al. Vulnerability to Low-Dose Combination of Irinotecan and Niraparib in ATM-Mutated Colorectal Cancer. J. Exp. Clin. Cancer Res. 2021, 40, 15.
- Lee, H.-J.; Choi, C.-H. Characterization of SN38-Resistant T47D Breast Cancer Cell Sublines Overexpressing BCRP, MRP1, MRP2, MRP3, and MRP4. BMC Cancer 2022, 22, 446.
- Goffart, S.; Hangas, A.; Pohjoismäki, J.L.O. Twist and Turn—Topoisomerase Functions in Mitochondrial DNA Maintenance. Int. J. Mol. Sci. 2019, 20, 2041.
- McLeod, H.L.; Keith, W.N. Variation in Topoisomerase I Gene Copy Number as a Mechanism for Intrinsic Drug Sensitivity. Br. J. Cancer 1996, 74, 508–512.
- McClendon, A.K.; Rodriguez, A.C.; Osheroff, N. Human Topoisomerase IIalpha Rapidly Relaxes Positively Supercoiled DNA: Implications for Enzyme Action Ahead of Replication Forks. J. Biol. Chem. 2005, 280, 39337–39345.
- Kumar, S.; Gahramanov, V.; Yaglom, J.; Patel, S.; Kaczmarczyk, L.; Alexandrov, I.; Gerlitz, G.; Salmon-Divon, M.; Sherman, M.Y. Homologous Recombination Repair Creates Mutations in the Non-Coding Genome That Alter Topoisomerase-1 Cleavage Sites & Orchestrates Irinotecan Resistance. bioRxiv 2022, 2021, 11.26.470089.
- Ju, B.-G.; Lunyak, V.V.; Perissi, V.; Garcia-Bassets, I.; Rose, D.W.; Glass, C.K.; Rosenfeld, M.G. A Topoisomerase IIbeta-Mediated DsDNA Break Required for Regulated Transcription. Science 2006, 312, 1798–1802.
- Baranello, L.; Wojtowicz, D.; Cui, K.; Devaiah, B.N.; Chung, H.-J.; Chan-Salis, K.Y.; Guha, R.; Wilson, K.; Zhang, X.; Zhang, H.; et al. RNA Polymerase II Regulates Topoisomerase 1 Activity to Favor Efficient Transcription. Cell 2016, 165, 357–371.
- Hsiang, Y.H.; Lihou, M.G.; Liu, L.F. Arrest of Replication Forks by Drug-Stabilized Topoisomerase I-DNA Cleavable Complexes as a Mechanism of Cell Killing by Camptothecin. Cancer Res. 1989, 49, 5077–5082.
- Zhao, H.; Rybak, P.; Dobrucki, J.; Traganos, F.; Darzynkiewicz, Z. Relationship of DNA Damage Signaling to DNA Replication Following Treatment with DNA Topoisomerase Inhibitors Camptothecin/Topotecan, Mitoxantrone, or Etoposide. Cytom. Part A 2012, 81A, 45–51.
- Saayman, X.; Graham, E.; Nathan, W.J.; Nussenzweig, A.; Esashi, F. Centromeres as Universal Hotspots of DNA Breakage, Driving RAD51-Mediated Recombination during Quiescence. Mol. Cell 2023, 83, 523–538.e7.
- D’Arpa, P.; Beardmore, C.; Liu, L.F. Involvement of Nucleic Acid Synthesis in Cell Killing Mechanisms of Topoisomerase Poisons1. Cancer Res. 1990, 50, 6919–6924.
- Lips, J.; Kaina, B. DNA Double-Strand Breaks Trigger Apoptosis in P53-Deficient Fibroblasts. Carcinogenesis 2001, 22, 579–585.
- Roos, W.P.; Kaina, B. DNA Damage-Induced Cell Death: From Specific DNA Lesions to the DNA Damage Response and Apoptosis. Cancer Lett. 2013, 332, 237–248.
- Rothenberg, M.L. Irinotecan (CPT-11): Recent Developments and Future Directions–Colorectal Cancer and Beyond. Oncologist 2001, 6, 66–80.
- Mathijssen, R.H.J.; Marsh, S.; Karlsson, M.O.; Xie, R.; Baker, S.D.; Verweij, J.; Sparreboom, A.; McLeod, H.L. Irinotecan Pathway Genotype Analysis to Predict Pharmacokinetics. Clin. Cancer Res. 2003, 9, 3246–3253.
- Fujita, K.; Kubota, Y.; Ishida, H.; Sasaki, Y. Irinotecan, a Key Chemotherapeutic Drug for Metastatic Colorectal Cancer. World J. Gastroenterol. 2015, 21, 12234–12248.
- Santos, A.; Zanetta, S.; Cresteil, T.; Deroussent, A.; Pein, F.; Raymond, E.; Vernillet, L.; Risse, M.L.; Boige, V.; Gouyette, A.; et al. Metabolism of Irinotecan (CPT-11) by CYP3A4 and CYP3A5 in Humans. Clin. Cancer Res. 2000, 6, 2012–2020.
- Kciuk, M.; Marciniak, B.; Kontek, R. Irinotecan—Still an Important Player in Cancer Chemotherapy: A Comprehensive Overview. Int. J. Mol. Sci. 2020, 21, 4919.
- Khageh Hosseini, S.; Kolterer, S.; Steiner, M.; von Manstein, V.; Gerlach, K.; Trojan, J.; Waidmann, O.; Zeuzem, S.; Schulze, J.O.; Hahn, S.; et al. Camptothecin and Its Analog SN-38, the Active Metabolite of Irinotecan, Inhibit Binding of the Transcriptional Regulator and Oncoprotein FUBP1 to Its DNA Target Sequence FUSE. Biochem. Pharmacol. 2017, 146, 53–62.
- Das, S.K.; Kuzin, V.; Cameron, D.P.; Sanford, S.; Jha, R.K.; Nie, Z.; Rosello, M.T.; Holewinski, R.; Andresson, T.; Wisniewski, J.; et al. MYC Assembles and Stimulates Topoisomerases 1 and 2 in a “Topoisome”. Mol. Cell 2022, 82, 140–158.e12.
- Arango, D.; Mariadason, J.M.; Wilson, A.J.; Yang, W.; Corner, G.A.; Nicholas, C.; Aranes, M.J.; Augenlicht, L.H. C-Myc Overexpression Sensitises Colon Cancer Cells to Camptothecin-Induced Apoptosis. Br. J. Cancer 2003, 89, 1757–1765.
- Wu, W.; Dong, J.; Gou, H.; Geng, R.; Yang, X.; Chen, D.; Xiang, B.; Zhang, Z.; Ren, S.; Chen, L.; et al. EGCG Synergizes the Therapeutic Effect of Irinotecan through Enhanced DNA Damage in Human Colorectal Cancer Cells. J. Cell. Mol. Med. 2021, 25, 7913–7921.
- Xu, R.-H.; Muro, K.; Morita, S.; Iwasa, S.; Han, S.W.; Wang, W.; Kotaka, M.; Nakamura, M.; Ahn, J.B.; Deng, Y.-H.; et al. Modified XELIRI (Capecitabine plus Irinotecan) versus FOLFIRI (Leucovorin, Fluorouracil, and Irinotecan), Both Either with or without Bevacizumab, as Second-Line Therapy for Metastatic Colorectal Cancer (AXEPT): A Multicentre, Open-Label, Randomised, Non-Inferiority, Phase 3 Trial. Lancet Oncol. 2018, 19, 660–671.
- Cunningham, D.; Humblet, Y.; Siena, S.; Khayat, D.; Bleiberg, H.; Santoro, A.; Bets, D.; Mueser, M.; Harstrick, A.; Verslype, C.; et al. Cetuximab Monotherapy and Cetuximab plus Irinotecan in Irinotecan-Refractory Metastatic Colorectal Cancer. N. Engl. J. Med. 2004, 351, 337–345.
- Zhang, X.; Duan, R.; Wang, Y.; Liu, X.; Zhang, W.; Zhu, X.; Chen, Z.; Shen, W.; He, Y.; Wang, H.Q.; et al. FOLFIRI (Folinic Acid, Fluorouracil, and Irinotecan) Increases Not Efficacy but Toxicity Compared with Single-Agent Irinotecan as a Second-Line Treatment in Metastatic Colorectal Cancer Patients: A Randomized Clinical Trial. Ther. Adv. Med. Oncol. 2022, 14, 17588359211068736.
- Fuchs, C.S.; Marshall, J.; Mitchell, E.; Wierzbicki, R.; Ganju, V.; Jeffery, M.; Schulz, J.; Richards, D.; Soufi-Mahjoubi, R.; Wang, B.; et al. Randomized, Controlled Trial of Irinotecan Plus Infusional, Bolus, or Oral Fluoropyrimidines in First-Line Treatment of Metastatic Colorectal Cancer: Results From the BICC-C Study. JCO 2007, 25, 4779–4786.
- Liu, X.; Ou, K.; Ma, X.; Gao, L.; Wang, Q.; Zhang, H.; Yang, L. Safety and Efficacy of Irinotecan, Oxaliplatin, and Capecitabine (XELOXIRI) Regimen with or without Targeted Drugs in Patients with Metastatic Colorectal Cancer: A Retrospective Cohort Study. BMC Cancer 2022, 22, 807.
- Wiseman, L.R.; Markham, A. Irinotecan. Drugs 1996, 52, 606–623.
- Porru, M.; Pompili, L.; Caruso, C.; Biroccio, A.; Leonetti, C. Targeting KRAS in Metastatic Colorectal Cancer: Current Strategies and Emerging Opportunities. J. Exp. Clin. Cancer Res. 2018, 37, 57.
- Fallik, D.; Borrini, F.; Boige, V.; Viguier, J.; Jacob, S.; Miquel, C.; Sabourin, J.-C.; Ducreux, M.; Praz, F. Microsatellite Instability Is a Predictive Factor of the Tumor Response to Irinotecan in Patients with Advanced Colorectal Cancer. Cancer Res. 2003, 63, 5738–5744.
- Bertagnolli, M.M.; Niedzwiecki, D.; Compton, C.C.; Hahn, H.P.; Hall, M.; Damas, B.; Jewell, S.D.; Mayer, R.J.; Goldberg, R.M.; Saltz, L.B.; et al. Microsatellite Instability Predicts Improved Response to Adjuvant Therapy with Irinotecan, Fluorouracil, and Leucovorin in Stage III Colon Cancer: Cancer and Leukemia Group B Protocol 89803. J. Clin. Oncol. 2009, 27, 1814–1821.
- Kawakami, H.; Zaanan, A.; Sinicrope, F.A. MSI Testing and Its Role in the Management of Colorectal Cancer. Curr. Treat. Options Oncol. 2015, 16, 30.
- Vilar, E.; Scaltriti, M.; Saura, C.; Guzman, M.; Macarulla, T.; Arribas, J.; Tabernero, J. Microsatellite Instability (MSI) Due to Mutation or Epigenetic Silencing Is Associated with Increased Cytotoxicity to Irinotecan (CPT-11) in Human Colorectal Cancer (CRC) Cell Lines. JCO 2007, 25, 10527.
- Evolution of resistance to Irinotecan in cancer cells involves generation of topoisomerase-guided mutations in non-coding genome that reduce the chances of DNA breaks . IJMS. Retrieved 2023-5-26
This entry is offline, you can click here to edit this entry!