Parkinson’s disease (PD) is a multifactorial disorder involving both motor and non-motor symptoms caused by the progressive death of distinct neuronal populations, including dopaminergic neurons in the substantia nigra. The deposition of aggregated α-synuclein protein into Lewy body inclusions is a hallmark of the disorder, and α-synuclein pathology has been found in the enteric nervous system (ENS) of PD patients up to two decades prior to diagnosis. In combination with the high occurrence of gastrointestinal dysfunction in early stages of PD, evidence strongly suggests that some forms of PD may originate in the gut.
- alpha-synuclein
- Parkinson’s disease
- enteric nervous system
1. Introduction
2. The Human Enteric Nervous System
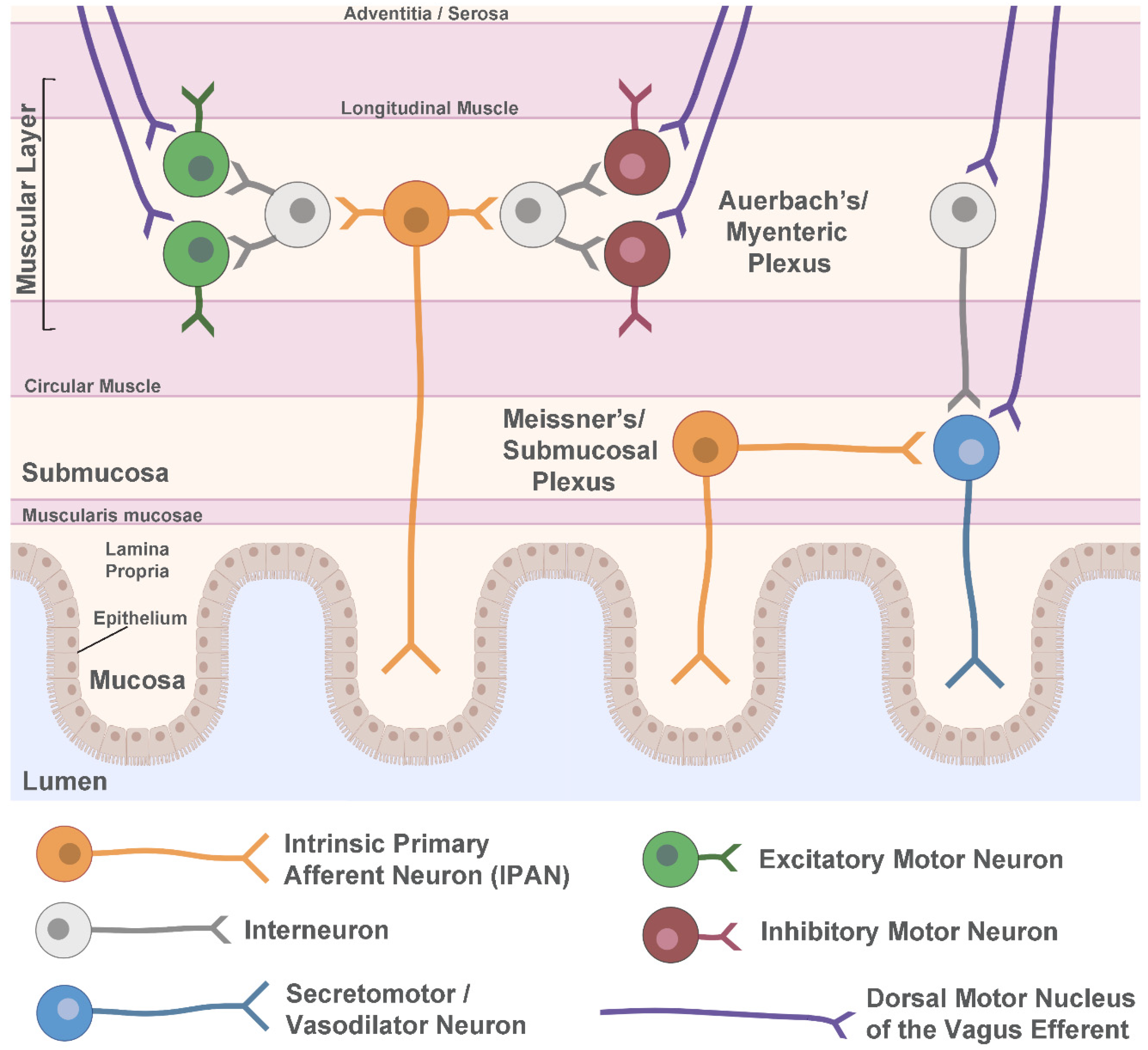
3. Lewy Pathology in the Enteric Nervous System in PD
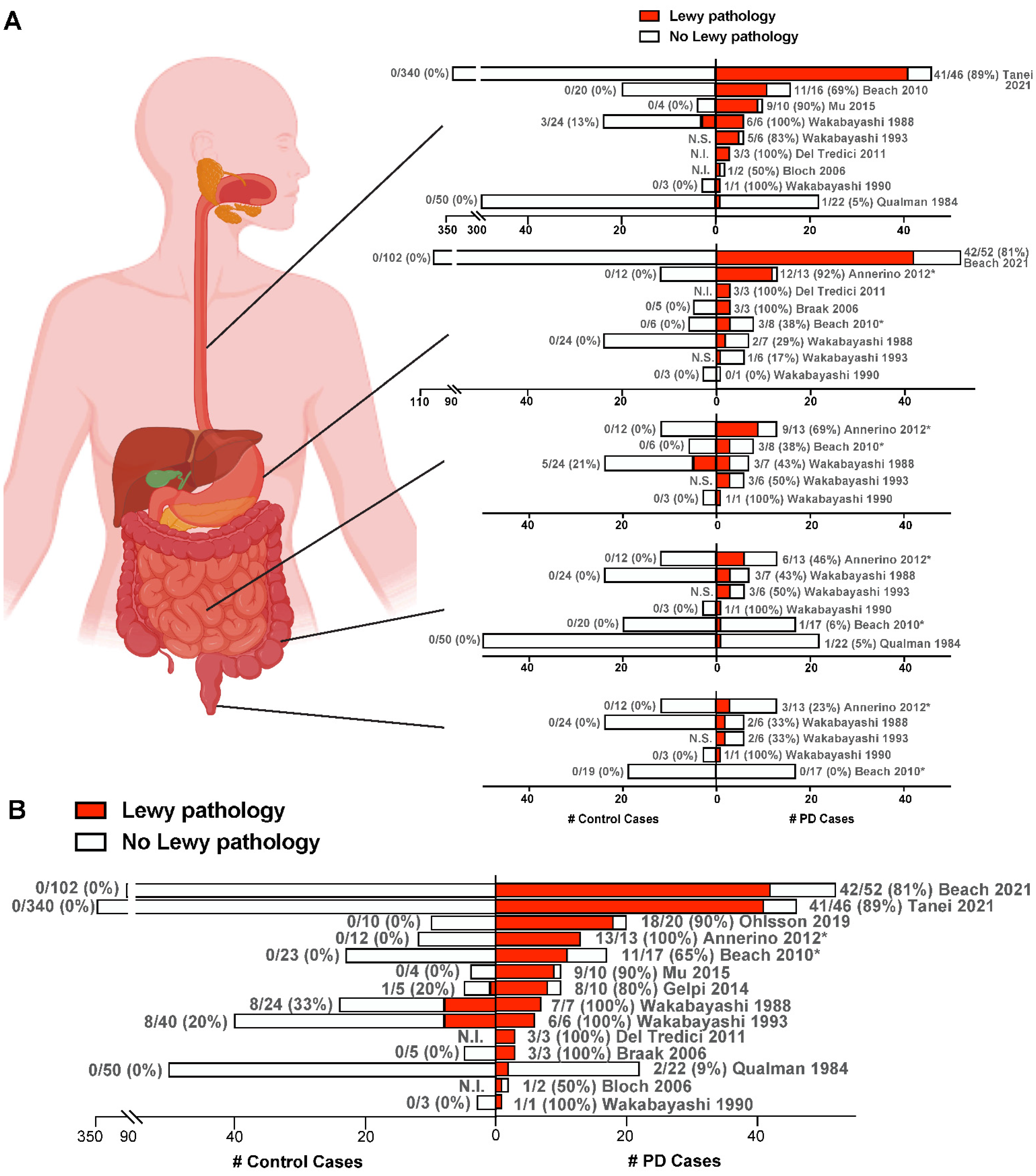
4. Does α-Synuclein Aggregation Begin in the Gut and Spread to the Brain?
4.1. Evidence from Incidental LB Disease and Prodromal PD
4.2. Evidence from Human Vagotomy Studies
4.3. Prion-Like Transmission of α-Synuclein in Rodents and Monkeys
4.4. C. elegans as a Powerful Model System to Study α-Synuclein Pathogenicity in PD
In an effort to generate prion-like α-synuclein transmission models initiated in the gut of C. elegans, a group recently published the neurotoxic effects of feeding worms human α-synuclein PFFs [71]. To our knowledge, this is the first report of α-synuclein PFF exposure in C. elegans. Similar to mouse models, scholars found that PFF ingestion in C. elegans promotes dopaminergic neurodegeneration, accelerates the aggregation of host α-synuclein in muscle, and induces an age-dependent motor decline. The development of these new models may serve to complement existing rodent model systems by acting as platforms for high-throughput discovery.
4.5. Alternative Hypotheses of α-Synuclein Spreading in PD
This entry is adapted from the peer-reviewed paper 10.3390/ijms24087205
References
- Ding, C.; Wu, Y.; Chen, X.; Chen, Y.; Wu, Z.; Lin, Z.; Kang, D.; Fang, W.; Chen, F. Global, regional, and national burden and attributable risk factors of neurological disorders: The Global Burden of Disease study 1990–2019. Front. Public Health 2022, 10, 952161.
- Forno, L.S. Neuropathology of Parkinson’s disease. J. Neuropathol. Exp. Neurol. 1996, 55, 259–272.
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha-synuclein in Lewy bodies. Nature 1997, 388, 839–840.
- Hoehn, M.M.; Yahr, M.D. Parkinsonism: Onset, progression and mortality. Neurology 1967, 17, 427–442.
- Lim, S.Y.; Fox, S.H.; Lang, A.E. Overview of the extranigral aspects of Parkinson disease. Arch. Neurol. 2009, 66, 167–172.
- Irwin, D.J.; Lee, V.M.; Trojanowski, J.Q. Parkinson’s disease dementia: Convergence of α-synuclein, tau and amyloid-β pathologies. Nature reviews. Neuroscience 2013, 14, 626–636.
- Fasano, A.; Visanji, N.P.; Liu, L.W.; Lang, A.E.; Pfeiffer, R.F. Gastrointestinal dysfunction in Parkinson’s disease. Lancet Neurol. 2015, 14, 625–639.
- Bullich, C.; Keshavarzian, A.; Garssen, J.; Kraneveld, A.; Perez-Pardo, P. Gut Vibes in Parkinson’s Disease: The Microbiota-Gut-Brain Axis. Mov. Disord. Clin. Pract. 2019, 6, 639–651.
- Qualman, S.J.; Haupt, H.M.; Yang, P.; Hamilton, S.R. Esophageal Lewy bodies associated with ganglion cell loss in achalasia. Similarity to Parkinson’s disease. Gastroenterology 1984, 87, 848–856.
- Kupsky, W.J.; Grimes, M.M.; Sweeting, J.; Bertsch, R.; Cote, L.J. Parkinson’s disease and megacolon: Concentric hyaline inclusions (Lewy bodies) in enteric ganglion cells. Neurology 1987, 37, 1253–1255.
- Wakabayashi, K.; Takahashi, H.; Takeda, S.; Ohama, E.; Ikuta, F. Parkinson’s disease: The presence of Lewy bodies in Auerbach’s and Meissner’s plexuses. Acta Neuropathol. 1988, 76, 217–221.
- Wakabayashi, K.; Takahashi, H.; Ohama, E.; Ikuta, F. Parkinson’s disease: An immunohistochemical study of Lewy body-containing neurons in the enteric nervous system. Acta Neuropathol. 1990, 79, 581–583.
- Wakabayashi, K.; Takahashi, H.; Ohama, E.; Takeda, S.; Ikuta, F. Lewy bodies in the visceral autonomic nervous system in Parkinson’s disease. Adv. Neurol. 1993, 60, 609–612.
- Braak, H.; de Vos, R.A.; Bohl, J.; Del Tredici, K. Gastric alpha-synuclein immunoreactive inclusions in Meissner’s and Auerbach’s plexuses in cases staged for Parkinson’s disease-related brain pathology. Neurosci. Lett. 2006, 396, 67–72.
- Bloch, A.; Probst, A.; Bissig, H.; Adams, H.; Tolnay, M. Alpha-synuclein pathology of the spinal and peripheral autonomic nervous system in neurologically unimpaired elderly subjects. Neuropathol. Appl. Neurobiol. 2006, 32, 284–295.
- Beach, T.G.; Adler, C.H.; Sue, L.I.; Vedders, L.; Lue, L.; White, C.L., III; Akiyama, H.; Caviness, J.N.; Shill, H.A.; Sabbagh, M.N.; et al. Arizona Parkinson’s Disease Consortium Multi-organ distribution of phosphorylated alpha-synuclein histopathology in subjects with Lewy body disorders. Acta Neuropathol. 2010, 119, 689–702.
- Del Tredici, K.; Duda, J.E. Peripheral Lewy body pathology in Parkinson’s disease and incidental Lewy body disease: Four cases. J. Neurol. Sci. 2011, 310, 100–106.
- Annerino, D.M.; Arshad, S.; Taylor, G.M.; Adler, C.H.; Beach, T.G.; Greene, J.G. Parkinson’s disease is not associated with gastrointestinal myenteric ganglion neuron loss. Acta Neuropathol. 2012, 124, 665–680.
- Gelpi, E.; Navarro-Otano, J.; Tolosa, E.; Gaig, C.; Compta, Y.; Rey, M.J.; Martí, M.J.; Hernández, I.; Valldeoriola, F.; Reñé, R.; et al. Multiple organ involvement by alpha-synuclein pathology in Lewy body disorders. Mov. Disord. Off. J. Mov. Disord. Soc. 2014, 29, 1010–1018.
- Mu, L.; Chen, J.; Sobotka, S.; Nyirenda, T.; Benson, B.; Gupta, F.; Sanders, I.; Adler, C.H.; Caviness, J.N.; Shill, H.A.; et al. Arizona Parkinson’s Disease Consortium Alpha-Synuclein Pathology in Sensory Nerve Terminals of the Upper Aerodigestive Tract of Parkinson’s Disease Patients. Dysphagia 2015, 30, 404–417.
- Beach, T.G.; Corbillé, A.G.; Letournel, F.; Kordower, J.H.; Kremer, T.; Munoz, D.G.; Intorcia, A.; Hentz, J.; Adler, C.H.; Sue, L.I.; et al. Multicenter Assessment of Immunohistochemical Methods for Pathological Alpha-Synuclein in Sigmoid Colon of Autopsied Parkinson’s Disease and Control Subjects. J. Park. Dis. 2016, 6, 761–770.
- Ohlsson, B.; Englund, E. Atrophic Myenteric and Submucosal Neurons Are Observed in Parkinson’s Disease. Park. Dis. 2019, 2019, 7935820.
- Tanei, Z.I.; Saito, Y.; Ito, S.; Matsubara, T.; Motoda, A.; Yamazaki, M.; Sakashita, Y.; Kawakami, I.; Ikemura, M.; Tanaka, S.; et al. Lewy pathology of the esophagus correlates with the progression of Lewy body disease: A Japanese cohort study of autopsy cases. Acta Neuropathol. 2021, 141, 25–37.
- Mayer, E.A.; Tillisch, K.; Gupta, A. Gut/brain axis and the microbiota. J. Clin. Investig. 2015, 125, 926–938.
- Petrov, V.A.; Saltykova, I.V.; Zhukova, I.A.; Alifirova, V.M.; Zhukova, N.G.; Dorofeeva, Y.B.; Tyakht, A.V.; Kovarsky, B.A.; Alekseev, D.G.; Kostryukova, E.S.; et al. Analysis of Gut Microbiota in Patients with Parkinson’s Disease. Bull. Exp. Biol. Med. 2017, 162, 734–737.
- Bedarf, J.R.; Hildebrand, F.; Coelho, L.P.; Sunagawa, S.; Bahram, M.; Goeser, F.; Bork, P.; Wüllner, U. Functional implications of microbial and viral gut metagenome changes in early stage L-DOPA-naïve Parkinson’s disease patients. Genome Med. 2017, 9, 39.
- Hasegawa, S.; Goto, S.; Tsuji, H.; Okuno, T.; Asahara, T.; Nomoto, K.; Shibata, A.; Fujisawa, Y.; Minato, T.; Okamoto, A.; et al. Intestinal Dysbiosis and Lowered Serum Lipopolysaccharide-Binding Protein in Parkinson’s Disease. PLoS ONE 2015, 10, e0142164.
- Unger, M.M.; Spiegel, J.; Dillmann, K.U.; Grundmann, D.; Philippeit, H.; Bürmann, J.; Faßbender, K.; Schwiertz, A.; Schäfer, K.H. Short chain fatty acids and gut microbiota differ between patients with Parkinson’s disease and age-matched controls. Park. Relat. Disord. 2016, 32, 66–72.
- Hill-Burns, E.M.; Debelius, J.W.; Morton, J.T.; Wissemann, W.T.; Lewis, M.R.; Wallen, Z.D.; Peddada, S.D.; Factor, S.A.; Molho, E.; Zabetian, C.P.; et al. Parkinson’s disease and Parkinson’s disease medications have distinct signatures of the gut microbiome. Mov. Disord. Off. J. Mov. Disord. Soc. 2017, 32, 739–749.
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047.
- Krüger, R.; Kuhn, W.; Müller, T.; Woitalla, D.; Graeber, M.; Kösel, S.; Przuntek, H.; Epplen, J.T.; Schöls, L.; Riess, O. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat. Genet. 1998, 18, 106–108.
- Zarranz, J.J.; Alegre, J.; Gómez-Esteban, J.C.; Lezcano, E.; Ros, R.; Ampuero, I.; Vidal, L.; Hoenicka, J.; Rodriguez, O.; Atarés, B.; et al. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann. Neurol. 2004, 55, 164–173.
- Proukakis, C.; Dudzik, C.G.; Brier, T.; MacKay, D.S.; Cooper, J.M.; Millhauser, G.L.; Houlden, H.; Schapira, A.H. A novel α-synuclein missense mutation in Parkinson disease. Neurology 2013, 80, 1062–1064.
- Lesage, S.; Anheim, M.; Letournel, F.; Bousset, L.; Honoré, A.; Rozas, N.; Pieri, L.; Madiona, K.; Dürr, A.; Melki, R.; et al. French Parkinson’s Disease Genetics Study Group G51D α-synuclein mutation causes a novel parkinsonian-pyramidal syndrome. Ann. Neurol. 2013, 73, 459–471.
- Pasanen, P.; Myllykangas, L.; Siitonen, M.; Raunio, A.; Kaakkola, S.; Lyytinen, J.; Tienari, P.J.; Pöyhönen, M.; Paetau, A. Novel α-synuclein mutation A53E associated with atypical multiple system atrophy and Parkinson’s disease-type pathology. Neurobiol. Aging 2014, 35, 2180.e1–2180.e5.
- Goedert, M.; Spillantini, M.G.; Del Tredici, K.; Braak, H. 100 years of Lewy pathology. Nat. Rev. Neurol. 2013, 9, 13–24.
- Braak, H.; Del Tredici, K.; Rüb, U.; de Vos, R.A.; Jansen Steur, E.N.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211.
- Fleming, M.A., 2nd; Ehsan, L.; Moore, S.R.; Levin, D.E. The Enteric Nervous System and Its Emerging Role as a Therapeutic Target. Gastroenterol. Res. Pract. 2020, 2020, 8024171.
- Gershon, M.D.; Margolis, K.G. The gut, its microbiome, and the brain: Connections and communications. J. Clin. Investig. 2021, 131, e143768.
- Chang, H.Y.; Mashimo, H.; Goyal, R.K. Musings on the wanderer: What’s new in our understanding of vago-vagal reflex? IV. Current concepts of vagal efferent projections to the gut. Am. J. Physiol. Gastrointest. Liver Physiol. 2003, 284, G357–G366.
- Benarroch, E.E. Enteric nervous system: Functional organization and neurologic implications. Neurology 2007, 69, 1953–1957.
- Mogor, F.; Kovács, T.; Lohinai, Z.; Dora, D. The Enteric Nervous System and the Microenvironment of the Gut: The Translational Aspects of the Microbiome-Gut-Brain Axis. Appl. Sci. 2021, 11, 12000.
- Hansen, C.; Angot, E.; Bergström, A.L.; Steiner, J.A.; Pieri, L.; Paul, G.; Outeiro, T.F.; Melki, R.; Kallunki, P.; Fog, K.; et al. α-Synuclein propagates from mouse brain to grafted dopaminergic neurons and seeds aggregation in cultured human cells. J. Clin. Investig. 2011, 121, 715–725.
- Desplats, P.; Lee, H.J.; Bae, E.J.; Patrick, C.; Rockenstein, E.; Crews, L.; Spencer, B.; Masliah, E.; Lee, S.J. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc. Natl. Acad. Sci. USA 2009, 106, 13010–13015.
- Li, J.Y.; Englund, E.; Holton, J.L.; Soulet, D.; Hagell, P.; Lees, A.J.; Lashley, T.; Quinn, N.P.; Rehncrona, S.; Björklund, A.; et al. Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat. Med. 2008, 14, 501–503.
- Kordower, J.H.; Chu, Y.; Hauser, R.A.; Freeman, T.B.; Olanow, C.W. Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson’s disease. Nat. Med. 2008, 14, 504–506.
- Peelaerts, W.; Bousset, L.; Van der Perren, A.; Moskalyuk, A.; Pulizzi, R.; Giugliano, M.; Van den Haute, C.; Melki, R.; Baekelandt, V. α-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature 2015, 522, 340–344.
- Recasens, A.; Dehay, B.; Bové, J.; Carballo-Carbajal, I.; Dovero, S.; Pérez-Villalba, A.; Fernagut, P.O.; Blesa, J.; Parent, A.; Perier, C.; et al. Lewy body extracts from Parkinson disease brains trigger α-synuclein pathology and neurodegeneration in mice and monkeys. Ann. Neurol. 2014, 75, 351–362.
- Watts, J.C.; Giles, K.; Oehler, A.; Middleton, L.; Dexter, D.T.; Gentleman, S.M.; DeArmond, S.J.; Prusiner, S.B. Transmission of multiple system atrophy prions to transgenic mice. Proc. Natl. Acad. Sci. USA 2013, 110, 19555–19560.
- Masuda-Suzukake, M.; Nonaka, T.; Hosokawa, M.; Oikawa, T.; Arai, T.; Akiyama, H.; Mann, D.M.; Hasegawa, M. Prion-like spreading of pathological α-synuclein in brain. Brain J. Neurol. 2013, 136 Pt 4, 1128–1138.
- Luk, K.C.; Kehm, V.M.; Zhang, B.; O’Brien, P.; Trojanowski, J.Q.; Lee, V.M. Intracerebral inoculation of pathological α-synuclein initiates a rapidly progressive neurodegenerative α-synucleinopathy in mice. J. Exp. Med. 2012, 209, 975–986.
- Luk, K.C.; Kehm, V.; Carroll, J.; Zhang, B.; O’Brien, P.; Trojanowski, J.Q.; Lee, V.M. Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 2012, 338, 949–953.
- Paumier, K.L.; Luk, K.C.; Manfredsson, F.P.; Kanaan, N.M.; Lipton, J.W.; Collier, T.J.; Steece-Collier, K.; Kemp, C.J.; Celano, S.; Schulz, E.; et al. Intrastriatal injection of pre-formed mouse α-synuclein fibrils into rats triggers α-synuclein pathology and bilateral nigrostriatal degeneration. Neurobiol. Dis. 2015, 82, 185–199.
- Corsi, A.K.; Wightman, B.; Chalfie, M. A Transparent Window into Biology: A Primer on Caenorhabditis Elegans. WormBook: The Online Review of C. Elegans Biology. 2015, pp. 1–31. Available online: http://www.wormbook.org/chapters/www_celegansintro/celegansintro.html (accessed on 20 February 2023).
- Kaletta, T.; Hengartner, M.O. Finding function in novel targets: C. elegans as a model organism. Nat. Rev. Drug Discov. 2006, 5, 387–398.
- Hobert, O. The Neuronal Genome of Caenorhabditis Elegans. WormBook: The Online Review of C. Elegans Biology. 2013, pp. 1–106. Available online: http://www.wormbook.org/chapters/www_neuronalgenome/neuronalgenome.html (accessed on 20 February 2023).
- Son, H.G.; Altintas, O.; Kim, E.J.E.; Kwon, S.; Lee, S.V. Age-dependent changes and biomarkers of aging in Caenorhabditis elegans. Aging Cell 2019, 18, e12853.
- Lakso, M.; Vartiainen, S.; Moilanen, A.M.; Sirviö, J.; Thomas, J.H.; Nass, R.; Blakely, R.D.; Wong, G. Dopaminergic neuronal loss and motor deficits in Caenorhabditis elegans overexpressing human alpha-synuclein. J. Neurochem. 2003, 86, 165–172.
- Van Ham, T.J.; Thijssen, K.L.; Breitling, R.; Hofstra, R.M.; Plasterk, R.H.; Nollen, E.A.C. C. elegans model identifies genetic modifiers of alpha-synuclein inclusion formation during aging. PLoS Genet. 2008, 4, e1000027.
- Cao, P.; Yuan, Y.; Pehek, E.A.; Moise, A.R.; Huang, Y.; Palczewski, K.; Feng, Z. Alpha-synuclein disrupted dopamine homeostasis leads to dopaminergic neuron degeneration in Caenorhabditis elegans. PLoS ONE 2010, 5, e9312.
- Mor, D.E.; Tsika, E.; Mazzulli, J.R.; Gould, N.S.; Kim, H.; Daniels, M.J.; Doshi, S.; Gupta, P.; Grossman, J.L.; Tan, V.X.; et al. Dopamine induces soluble α-synuclein oligomers and nigrostriatal degeneration. Nat. Neurosci. 2017, 20, 1560–1568.
- Mor, D.E.; Sohrabi, S.; Kaletsky, R.; Keyes, W.; Tartici, A.; Kalia, V.; Miller, G.W.; Murphy, C.T. Metformin rescues Parkinson’s disease phenotypes caused by hyperactive mitochondria. Proc. Natl. Acad. Sci. USA 2020, 117, 26438–26447.
- Mor, D.E.; Murphy, C.T. Mitochondrial hyperactivity as a potential therapeutic target in Parkinson’s disease. Transl. Med. Aging 2020, 4, 117–120.
- Beach, T.G.; Adler, C.H.; Sue, L.I.; Shill, H.A.; Driver-Dunckley, E.; Mehta, S.H.; Intorcia, A.J.; Glass, M.J.; Walker, J.E.; Arce, R.; et al. Vagus Nerve and Stomach Synucleinopathy in Parkinson’s Disease, Incidental Lewy Body Disease, and Normal Elderly Subjects: Evidence Against the “Body-First” Hypothesis. J. Park. Dis. 2021, 11, 1833–1843.
- Lakso, M.; Vartiainen, S.; Moilanen, A.M.; Sirviö, J.; Thomas, J.H.; Nass, R.; Blakely, R.D.; Wong, G. Dopaminergic neuronal loss and motor deficits in Caenorhabditis elegans overexpressing human alpha-synuclein. J. Neurochem. 2003, 86, 165–172.
- Van Ham, T.J.; Thijssen, K.L.; Breitling, R.; Hofstra, R.M.; Plasterk, R.H.; Nollen, E.A. C. elegans model identifies genetic modifiers of alpha-synuclein inclusion formation during aging. PLoS Genet. 2008, 4, e1000027.
- Cao, P.; Yuan, Y.; Pehek, E.A.; Moise, A.R.; Huang, Y.; Palczewski, K.; Feng, Z. Alpha-synuclein disrupted dopamine homeostasis leads to dopaminergic neuron degeneration in Caenorhabditis elegans. PLoS ONE 2010, 5, e9312.
- Mor, D.E.; Tsika, E.; Mazzulli, J.R.; Gould, N.S.; Kim, H.; Daniels, M.J.; Doshi, S.; Gupta, P.; Grossman, J.L.; Tan, V.X.; et al. Dopamine induces soluble alpha-synuclein oligomers and nigrostriatal degeneration. Nat. Neurosci. 2017, 20, 1560–1568.
- Mor, D.E.; Sohrabi, S.; Kaletsky, R.; Keyes, W.; Tartici, A.; Kalia, V.; Miller, G.W.; Murphy, C.T. Metformin rescues Parkinson’s disease phenotypes caused by hyperactive mitochondria. Proc. Natl. Acad. Sci. USA 2020, 117, 26438–26447.
- Mor, D.E.; Murphy, C.T. Mitochondrial hyperactivity as a potential therapeutic target in Parkinson’s disease. Transl. Med. Aging 2020, 4, 117–120.
- Chen, M.; Vincent, J.; Ezeanii, A.; Wakade, S.; Yerigenahally, S.; Mor, D.E. Heparan sulfate proteoglycans mediate prion-like alpha-synuclein toxicity in Parkinson’s in vivo models. Life Sci. Alliance 2022, 5, e202201366.