Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
ABCG2 is an efflux transporter responsible for inducing multidrug resistance (MDR) in leukemic cells; through its ability to extrude many antineoplastic drugs, it leads to AML resistance and/or relapse. Moreover, ABCG2 may be co-expressed with other MDR-related proteins and is finely regulated by epigenetic mechanisms.
- acute myeloid leukemia
- multidrug resistance
- ABCG2
1. Introduction
Acute myeloid leukemia (AML) is an aggressive heterogenous disease arising from the accumulation and clonal expansion of somatic-driven mutations in CD34+/CD38- hematopoietic progenitors, which demonstrate increased proliferation, survival, and impaired maturation capacity [1][2]. It is the most common form of acute leukemia in adults, with a median age at diagnosis of 68 years and a sharp increase in incidence in the following decades [3]. In addition, AML prevalence is increased by therapy-related AML, which accounts for 10–15% of newly diagnosed AML, an effect of improved survival after anti-cancer therapies [4]. The estimated 5-year overall survival (OS) of AML is around 30%, with great differences between age groups (≈50% in younger patients and only ≈10% in elderly patients) and with disappointing progress over the past five decades, particularly in the older population [5][6].
Despite this, the occurrence of intrinsic or acquired drug resistance still results in AML being an aggressive disease and a major challenge for clinicians. Clinical multidrug resistance (MDR) is a multi-factorial phenomenon depending on host variations, drug–drug interaction, deregulation of cell death mechanisms, failure of DNA damage response and repair, epigenetic alterations, intratumor heterogeneity, and microenvironment alteration. MDR protects leukemia cell from immune surveillance and from the alteration in intracellular drug concentrations due to overexpression of membrane drug transporter proteins [7]. Acquired MDR has been intensively studied, and the molecular basis of this phenomenon is well established [8]. The cross-resistance to different, structurally unrelated anti-cancer drugs, such as vinblastine, vincristine, and daunomycin, developed by Chinese hamster lung cells grown in actinomycin D to select resistant cells, was first described more than 50 years ago [9]. A few years later, another study reported that daunomycin was actively transported out of multidrug-resistant cells [10]. The hypothesis of a promiscuous membrane transporter able to confer MDR was confirmed by the identification in Chinese hamster ovary cells of the “P-glycoprotein”, so called for the altered membrane permeability associated with its expression in resistant cells [11] and by characterization of its encoding gene [12]. The human homologue gene was soon identified and referred to as ATP-binding cassette (ABC) subfamily B1, ABCB1 [13]. Its recognition paved the way to studies of ABC transporters, leading to the identification of 48 human membrane proteins, grouped into 7 subfamilies, involved in different physiological biochemical and developmental processes beyond cancer drug transport [14][15]. The ABC superfamily is highly conserved among plant and animal species, mainly acting as import pumps in prokaryotes [16]. In eukaryotic cells, they act as exporters, pumping out substances from the cytoplasm or entrapping them into intracellular organelles, such as peroxisome, endoplasmic reticulum, or lysosomes, using an energy-dependent process involving binding and hydrolysis of ATP [17][18].
The general architecture of ABC proteins consists of two cytoplasmic nucleotide-binding domains (NBDs), which bind and hydrolyze ATP, and of two sets of hydrophobic transmembrane domains (TMDs), which transport substrates. ABC genes encode either a full transporter or a half transporter with a single TMD domain and a single NBD. Half transporters must dimerize as either homo- or heterodimers to form an active protein [15]. Whereas the structure and function of NBDs are similar throughout ABC subfamilies, TMDs are highly heterogeneous, thus permitting the recognition of different substrates and their translocation across membranes, irrespective of concentration gradient [19]. Despite the huge amount of data on ABC structure obtained by electron microscopy, the precise translocation mechanism remains elusive. It should be underlined that among the 48 ABC members, some have “narrow” substrate specificity, while others (19 of the 48) have broad specificity and are able to transport a wide range of anticancer drugs and to cause drug resistance if overexpressed in tumor cells [20]. The study of “null” mutants established via germline mutations has underscored the diversity of their physiological role and the consequences of their dysfunction. So, at present, more than 20 ABC proteins, belonging to all the identified sub-families, have been associated with human diseases [15][17]. The most important steps in ABC family discoveries are reported in Figure 1.
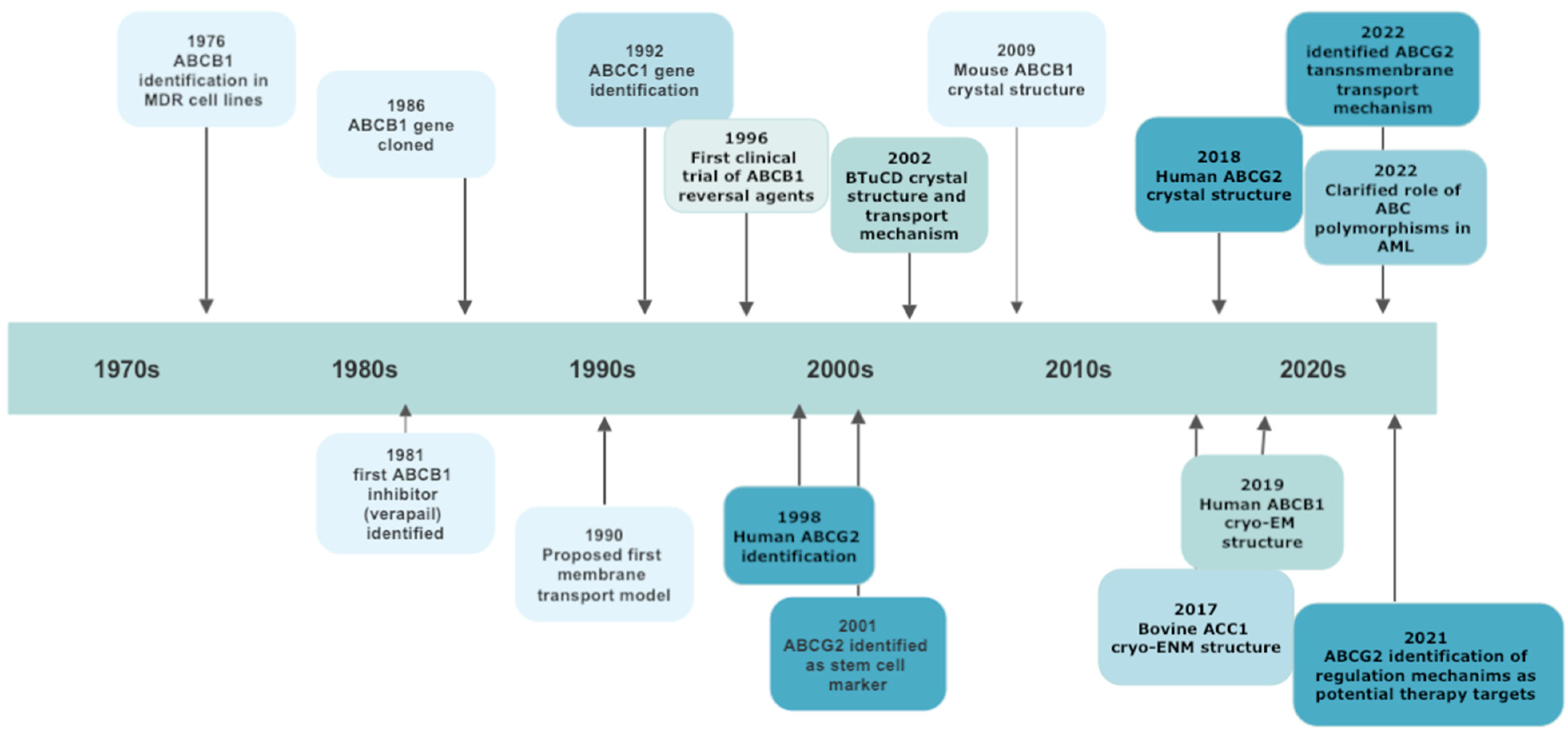
Figure 1. A schematic chronology of significant discoveries of ABC transporters involved in multidrug resistance.
2. ABCG Subfamily
2.1. ABCG1
ABCG1 is coded on chromosome 21q.22.3 and contributes to cholesterol transport and to cellular cholesterol homeostasis [21]. The protein is expressed in many cell types, including endothelial cells, lymphocytes, and myeloid cells on the cell membrane and in endosomes [22]. ACBG1 seems to regulate T cell development in the thymus by regulating intracellular cholesterol levels [23][24]. Moreover, ABCG1 is involved in innate immune response by the regulation of inflammation via reduction of inflammatory cytokines, and in anti-tumor immunity by favoring IL-4–mediated macrophage M2 polarization, producing a pro-cancer effect [25]. Moreover, it seems to be upregulated in lung cancer tissue, and aberrant expression of ABCG1 in lung cancer cells promotes proliferation, migration, and tumor invasion [26].
2.2. ABCG4
ABCG4 is mainly localized in the central nervous system (CNS) and seems to have a protective role in Alzheimer’s disease through inhibitory effects on amyloid β production [27]. Furthermore, ABCG4 overexpression seems to confer drug resistance, despite the mechanism not being completely understood [28].
2.3. ABCG5 and ABCG8
ABCG5 and ABCG8 form an obligated heterodimeric complex (G5/G8), highly expressed on epithelial cells of the intestine and liver, where it mediates sterol transport [29]. Gene variations are associated with hypercholesterolemia, platelet dysfunction, sitosterolemia, cardiovascular disease, and gallstones [30][31][32].
2.4. ABCG2
ABCG2 is the most studied among ABCG members. In the early 1990s, the observation of MDR in cell lines selected with mitoxantrone lacking MDR1 and MRP1 expression led to identification of a new transporter protein. The gene responsible for the novel resistance phenotype, first cloned by Doyle et al., in the MCF-7 Adr/Vp cell line, was named BCRP for breast cancer resistance protein, from the cell line origin [33][34][35], and further designed as ABCG2 by the Human Genome Organization Committee. The ABCG2 gene is highly conserved among species, most of which have a single gene present [36]. The exceptions are rodent and fish, which have more ABCG2 genes [37]. The human ABCG2 gene is located on chromosome 4, band 4q21–4q22, and extend over 66 kb containing 16 exons (range size from 60 to 532 bp) and 15 introns. The translational start site is on exon 2, ABC signature motif in exon 6, and the ATP binding sites (Walker motif A and B) in exon 3 and exon 6. The promoter region is located approximately 312 bp from the transcriptional start site [38].
In vitro studies demonstrated the upregulation of the ABCG2 gene under hypoxic conditions by estradiol progesterone and by aryl hydrocarbon receptor agonists [39][40][41][42]. ABCG2 expression can also be induced via peroxisome proliferator-activated receptor γ (PPARγ) [43] and downregulated by dexamethasone via the glucocorticoid receptor (GR) [44]. However, data on ABCG2 regulation are often controversial, and it has been hypothesized that the observed contradiction may be due to cell- or organ-specific regulation. Epigenetic regulation has been observed in overexpressing cell lines, where elevated ABCG2 levels were associated with hypomethylation or unmethylation of the CpG island and with hyperacetylation of the ABCG2 promoter [45].
At least one dimerization is required to produce a functional protein; dimerization is under the control of the endoplasmic reticulum quality control (ERQC) network. If passing the ERQC, protein traffics to the Golgi apparatus, where the protein becomes fully glycosylated, passes a further quality control, and can be delivered to its destination, the plasma membrane. Mechanisms by which the protein is transferred to the cell membrane are not completely elucidated and may include direct delivery, trafficking via the endosomal pool or trans-cytotic pathway via the basolateral membrane. Surplus protein is proteolytically degraded in lysosomes, and misfolded proteins become ubiquitinated and degraded in proteosome. Interestingly, 40–60% of the produced protein, even in wild form, does not pass ERQC and is eliminated [46]. Differently to ABCG5/ABCG8, ABCG2 may work only as homodimer; however, several studies have reported that it can assemble in higher oligomeric forms, from tetramers to dodecamers, a process sometimes favored by the presence of single-nucleotide polymorphisms [46][47].
2.5. ABCG2 Substrates
Since ABCG2 was first described, the list of its substrates has been steadily expanding. The substrate specificity of ABCG2 is highly overlapping to that of ABCB1, and like ABCB1, ABCG2 preferentially targets hydrophobic and lipophilic compounds with planar aromatic systems. Transfer across the membrane is associated with conformation changes of the protein (“in-facing”, with the substrate binding site open in the cytoplasm, and “out-facing” open in the extracellular space). Recently, Gyöngy et al., and Yu et al., proposed two molecular models to explain drug transport, highlighting the crucial role of ATP binding to modulate ABCG2 conformation [48][49]. As for the other ABC members, the molecular basis of ABCG2 substrate specificity is not fully elucidated. It has been hypothesized that substrate binding depends on the formation of a “membrane entrance” in the lipid bilayer by hydrophobic amino-acid residuals, available in the “in-facing” protein conformation, and that the different combination of these residuals provides the substrate specificity [50].
Mitoxantrone transport is the hallmark of the cells expressing ABCG2; thus, the first recognized substrates were predominantly chemotherapy agents. More recently, other classes of substrates have been identified, including antivirals, HMG CoA inhibitors, flavonoids, carcinogens, and calcium channel blockers [51][52].
3. Expression and Clinical Significance of ABCG2 in AML
Despite the role of ABC proteins in determining drug resistance in hematologic and solid cancers, it is still a matter of controversy, and many studies in the past decades have shown a relationship between ABCG2 overexpression and poor clinical outcome in AML. The heterogeneity of the employed methods, including ABCG2 m-RNA expression, protein evaluation by flow cytometry or immune-cytochemistry, ABCG2 efflux of fluorescent substrates, and the lack of standardization may, in part, explain the confounding results and make data comparison difficult.
In 2001, Sargent et al., evaluated ABCG2 expression by immunocytochemistry, using the anti ABCG2 BXP-34 monoclonal antibody, in 20 samples of de novo AML (12 previously treated and 8 untreated). ABCG2 positivity showed high variability, but 27% patients had more than 10% positive cells and were considered as ABCG2 overexpressing. There were no differences between pretreated and naïve patients with regard to FAB cytotype or other clinical/biological characteristics.
Van den Heuvel-Eibrink et al., compared ABCB1, ABCC1, LRP, and ABCG2 mRNA levels in 20 leukemia samples at diagnosis or relapse, observing significantly higher ABCG2 mRNA levels at relapse (median 1.7-fold, p = 0.04). On the contrary, expression levels of the other tested ABC proteins did not change. They hypothesized that only ABCG2 accounted for drug resistance at relapse [53]. In 2007, the same group explored the relevance of the same genes’ (ABCB1, ABCC1, LRP, and ABCG2) expression in a cohort of 154 elderly patients, observing a negative correlation between ABCB1 and ABCG2 expression and WBC count (p = 0.001) and a positive association between ABCB1 and ABCG2 and CD34 blast expression (p = 0.001). Moreover, high ABCB1/ABCG2 expression significantly reduced complete remission (CR) rate (p = 0.03) and seemed to be associated with a reduced event-free survival (EFS) (p = 0.05) [54].
The prognostic significance of ABCG2 expression in pediatric AML was assessed by Steinbach et al., The authors evaluated ABCG2 expression by real time PCR in 59 pediatric cases, reporting 10-fold higher mRNA levels in patients not in remission after induction therapy compared to those achieving CR (p = 0.012). The highest expression was observed in the M1-M2 FAB subtype and the lowest in M5 (p = 0.004). Worse OS was reported in ABCG2 overexpressing cases, irrespective of disease risk (p = 0.023) [55]. The same group also investigated the prognostic relevance of the co-expression of ABCB1, ABCG2, ABCC3, and ABCA3 in 112 children with AML treated according to the AML-BFM 2004 protocol. Patients with high levels of ABCG2 and ABCC3 had reduced CR rate, and those with ABCG2 overexpression also had lower DFS. As for CR probability in adults (Liu et al. [56]), DFS in this pediatric cohort was negatively affected by the number of overexpressed ABC transporters (p < 0.001) [57].
Taken together, this data supports a role of ABCG2 in affecting AML outcome, both in adults and in children. Poor response to induction therapy may be also influenced by the frequent co-expression of ABCB1, but the stem cell–like properties conferred to leukemic cells by ABCG2 overexpression may account for high relapse rate and for poor survival when induction therapy is intensified or despite allogeneic stem cell transplantation, even if performed in CR.
4. ABCG2 Polymorphisms in AML
The cloning of ABCG2 DNA from drug-selected cell lines and from normal tissues revealed many amino-acid substitutions able to alter the protein function and the substrate preference. Moreover, mutational analysis identified more than 80 ethnic-associated synonymous and non-synonymous single-nucleotide polymorphisms (SNPs) potentially influencing ABCG2 expression and function and affecting drug absorption, plasma concentration, and distribution and elimination (Figure 1) [58].
In drug-selected cell lines such as S1-M1-80 and MCF7/AdVp3000, unique mutations in amino acid position 482 makes cells highly resistant to mitoxantrone and doxorubicin. The replacement of Arg with Gly or Thr at position 482 increases rhodamine and anthracycline efflux compared to the wild-type counterpart [59]. In contrast, the R482G and R482T variants negatively affect ABCG2’s ability to transport methotrexate but confer increased methotrexate resistance [60]. At least 13 ABCG2 variants with substitution at R482 have been described in cell lines, all associated with strong resistance to mitoxantrone. The COO- terminus of TMD3, near position 482, and 3D homology models suggest that R482 is the central cavity of the binding pocket, with a crucial function in drug transmembrane translocation [61]. Mutations at N557 and H630 confer lower resistance to SN-38, although mitoxantrone resistance is maintained [62]. Mutations at C603 may impair the homodimer formation [47], and substitutions at N596 may affect N-linked glycosylation, reducing the amount of ABCG2 on the cell membrane [63]. Among synonymous SNPs, Q141K has been associated with increased risk of gout in Asian [64] and American populations [65].
5. ABCG2 and Extracellular Vesicles
Among the factors mediating the acquisition of the MDR phenotype, the role of intercellular communications has recently emerged. In this setting, extracellular vesicles (EVs) play a pivotal role [66]. EVs are a heterogenous group of lipid bilayer structures derived from either endosomal multivesicular bodies (exosomes) or from the plasma membrane (micro vesicles, also called ectosomes). Cells that secrete more vesicles show a greater level of resistance [67]. Many studies have shown that cytotoxic drugs may be sequestered into EVs and released from the cells, thus preventing their accumulation in the nucleus. This mechanism has been demonstrated in MCF7 breast cancer–resistant cells, in which ABCG2 efflux protein localizes to EVs and mediates the uptake of drugs in the vesicles before their release [68]. The compartmentalization of ABCG2 in EVs and not in other intracellular compartments depends on the activation of the PI3K/AKT pathway, suggesting a potential therapeutic target to overcome drug resistance by the inhibition of EVs biogenesis. Preliminary in vitro studies using the specific PI3K-AKT axis results in the reduction of EV number and volume. ABCG2 is relocated to the intracellular compartment and loses the ability to concentrate anticancer drugs, thus restoring cell sensitivity [68]. EVs may act also by transferring between cells the efflux transporters or mi-RNAs involved in the regulation of efflux proteins’ expression [69].
6. ABCG2 Inhibition
Different approaches to overcome ABCG2-mediated MDR have been proposed. Despite some positive results obtained in preventing ABCB1 binding [70][71], the attempt to develop chemotherapy agents that are not recognized by ABCG2 remains challenging, due to the molecular variety of transported compounds and the still incomplete knowledge of binding mechanisms [72]. The attractive strategy to induce collateral sensitivity (CS), a well-known phenomenon in which, due to overexpression of ABC transporters, a MDR cell becomes hypersensitive to some unrelated anticancer drugs, is difficult to pursue due to the variety of mechanisms involved in drug resistance [73].
So far, the most used method remains an inhibition of the efflux function, re-sensitizing resistant cells to conventional anticancer drugs. The first identified functional inhibitor of ABCG2 was fumitremorgin C (FTC), a mycotoxin produced by Aspergillus fumigatus, extensively used in experimental settings to test drug sensitivity in ABCG2 overexpressing cells [74].
It must be underlined that many of the molecules with inhibitory activity work only at high concentrations, often not attainable in clinical use. Among novel molecules, Ko143 (an FTC derivative), chromone derivatives, and a recently discovered indenoindol derivative are the most promising agents, having an IC50 in the nanomolar range [75]. Many ABCG2 inhibitors, like calcium channel blockers, anti-HIV drugs, and xanthine-oxidase inhibitors, were selected by drug repurposing, with the potential advantage of shortening the drug development process. The most interesting are tivozanib, fostamatinib, ponatinib, and febuxostat, all active at nanomolar IC50 [76][77][78][79].
Other new anticancer drugs have recently demonstrated an ABCG2 inhibitory effect in vitro. Sorf et al., demonstrated that ribociclib, a C4C/6 inhibitor approved for the treatment of locally advanced/metastatic breast cancer, inhibits ABCB1- and ABCG2-mediated daunorubicin and mitoxantrone efflux in AML cell lines at IC50 ranging between 1,4–3 μM, suggesting that combination therapy can revert MDR, especially in CD34+/FLT3-WT cases [80]. In vitro inhibition of ABCG2- and ABCC1-mediated daunorubicin and mitoxantrone efflux was demonstrated also by talazoparib, a drug approved for metastatic BCRA1/2 mutated breast cancer [81].
Despite the discovery of several molecules with an inhibitory effect, the interest in the clinical development of effective ABCG2 inhibitors has been deprioritized, likely due to the disappointing results attained in ABCB1 inhibition, and at present no clinical trials including ABCG2 inhibitors are ongoing. This is in contrast with the evident contribution of ABC transporters in chemotherapy failure, at least in AML. In this setting, ABCG2 overexpression not only accounts for an increased relapse risk and poor survival after conventional therapy, but also identifies a subset of patients at higher risk of relapse after allogeneic transplantation, which is still recognized as the only “curative” option for high-risk disease [82]. It is possible that the acquisition by ABCG2-overexpressing leukemic cells of stem-cell-like properties eventually favors their survival in transplant preparative regimens and their escape from post-transplant graft versus the leukemia effect. On this basis, strategies to counteract ABCG2 should be adopted.
This entry is adapted from the peer-reviewed paper 10.3390/ijms24087147
References
- Huntly, B.J.; Gilliland, D.G. Leukaemia stem cells and the evolution of cancer-stem-cell research. Nat. Rev. Cancer 2005, 5, 311–321.
- Jan, M.; Snyder, T.M.; Corces-Zimmerman, M.R.; Vyas, P.; Weissman, I.L.; Quake, S.R.; Majeti, R. Clonal evolution of preleukemic hematopoietic stem cells precedes human acute myeloid leukemia. Sci. Transl. Med. 2012, 4, 149ra118.
- Juliusson, G.; Antunovic, P.; Derolf, A.; Lehmann, S.; Mollgard, L.; Stockelberg, D.; Tidefelt, U.; Wahlin, A.; Hoglund, M. Age and acute myeloid leukemia: Real world data on decision to treat and outcomes from the Swedish Acute Leukemia Registry. Blood 2009, 113, 4179–4187.
- Morton, L.M.; Dores, G.M.; Schonfeld, S.J.; Linet, M.S.; Sigel, B.S.; Lam, C.J.K.; Tucker, M.A.; Curtis, R.E. Association of Chemotherapy for Solid Tumors With Development of Therapy-Related Myelodysplastic Syndrome or Acute Myeloid Leukemia in the Modern Era. JAMA Oncol. 2019, 5, 318–325.
- Sasaki, K.; Ravandi, F.; Kadia, T.M.; DiNardo, C.D.; Short, N.J.; Borthakur, G.; Jabbour, E.; Kantarjian, H.M. De novo acute myeloid leukemia: A population-based study of outcome in the United States based on the Surveillance, Epidemiology, and End Results (SEER) database, 1980 to 2017. Cancer 2021, 127, 2049–2061.
- Kantarjian, H.M.; Short, N.J.; Fathi, A.T.; Marcucci, G.; Ravandi, F.; Tallman, M.; Wang, E.S.; Wei, A.H. Acute Myeloid Leukemia: Historical Perspective and Progress in Research and Therapy Over 5 Decades. Clin. Lymphoma Myeloma Leuk. 2021, 21, 580–597.
- Assaraf, Y.G.; Brozovic, A.; Goncalves, A.C.; Jurkovicova, D.; Line, A.; Machuqueiro, M.; Saponara, S.; Sarmento-Ribeiro, A.B.; Xavier, C.P.R.; Vasconcelos, M.H. The multi-factorial nature of clinical multidrug resistance in cancer. Drug Resist. Updat 2019, 46, 100645.
- Gottesman, M.M.; Lavi, O.; Hall, M.D.; Gillet, J.P. Toward a Better Understanding of the Complexity of Cancer Drug Resistance. Annu. Rev. Pharmacol. Toxicol. 2016, 56, 85–102.
- Biedler, J.L.; Riehm, H. Cellular resistance to actinomycin D in Chinese hamster cells in vitro: Cross-resistance, radioautographic, and cytogenetic studies. Cancer Res. 1970, 30, 1174–1184.
- Dano, K. Active outward transport of daunomycin in resistant Ehrlich ascites tumor cells. Biochim. Et Biophys. Acta (BBA)-Biomembr. 1973, 323, 466–483.
- Juliano, R.L.; Ling, V. A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. Biochim. Et Biophys. Acta (BBA)-Biomembr. 1976, 455, 152–162.
- Gros, P.; Croop, J.; Roninson, I.; Varshavsky, A.; Housman, D.E. Isolation and characterization of DNA sequences amplified in multidrug-resistant hamster cells. Proc. Natl. Acad. Sci. USA 1986, 83, 337–341.
- Roninson, I.B.; Chin, J.E.; Choi, K.G.; Gros, P.; Housman, D.E.; Fojo, A.; Shen, D.W.; Gottesman, M.M.; Pastan, I. Isolation of human mdr DNA sequences amplified in multidrug-resistant KB carcinoma cells. Proc. Natl. Acad. Sci. USA 1986, 83, 4538–4542.
- Gottesman, M.M.; Ling, V. The molecular basis of multidrug resistance in cancer: The early years of P-glycoprotein research. FEBS Lett. 2006, 580, 998–1009.
- Theodoulou, F.L.; Kerr, I.D. ABC transporter research: Going strong 40 years on. Biochem. Soc. Trans. 2015, 43, 1033–1040.
- Higgins, C.F. ABC transporters: From microorganisms to man. Annu. Rev. Cell Biol. 1992, 8, 67–113.
- Dean, M.; Annilo, T. Evolution of the ATP-binding cassette (ABC) transporter superfamily in vertebrates. Annu. Rev. Genomics. Hum. Genet. 2005, 6, 123–142.
- Patel, H.; Wu, Z.X.; Chen, Y.; Bo, L.; Chen, Z.S. Drug resistance: From bacteria to cancer. Mol. Biomed. 2021, 2, 27.
- Dean, M.; Hamon, Y.; Chimini, G. The human ATP-binding cassette (ABC) transporter superfamily. J. Lipid Res. 2001, 42, 1007–1017.
- Fletcher, J.I.; Williams, R.T.; Henderson, M.J.; Norris, M.D.; Haber, M. ABC transporters as mediators of drug resistance and contributors to cancer cell biology. Drug Resist. Updat. 2016, 26, 1–9.
- Liang, X.; Wang, C.; Sun, Y.; Song, W.; Lin, J.; Li, J.; Guan, X. p62/mTOR/LXRalpha pathway inhibits cholesterol efflux mediated by ABCA1 and ABCG1 during autophagy blockage. Biochem. Biophys. Res. Commun. 2019, 514, 1093–1100.
- Sag, D.; Cekic, C.; Wu, R.; Linden, J.; Hedrick, C.C. The cholesterol transporter ABCG1 links cholesterol homeostasis and tumour immunity. Nat. Commun. 2015, 6, 6354.
- Armstrong, A.J.; Gebre, A.K.; Parks, J.S.; Hedrick, C.C. ATP-binding cassette transporter G1 negatively regulates thymocyte and peripheral lymphocyte proliferation. J. Immunol. 2010, 184, 173–183.
- Wang, F.; Beck-Garcia, K.; Zorzin, C.; Schamel, W.W.; Davis, M.M. Inhibition of T cell receptor signaling by cholesterol sulfate, a naturally occurring derivative of membrane cholesterol. Nat. Immunol. 2016, 17, 844–850.
- Goossens, P.; Rodriguez-Vita, J.; Etzerodt, A.; Masse, M.; Rastoin, O.; Gouirand, V.; Ulas, T.; Papantonopoulou, O.; Van Eck, M.; Auphan-Anezin, N.; et al. Membrane Cholesterol Efflux Drives Tumor-Associated Macrophage Reprogramming and Tumor Progression. Cell Metab. 2019, 29, 1376–1389.e1374.
- Tian, C.; Huang, D.; Yu, Y.; Zhang, J.; Fang, Q.; Xie, C. ABCG1 as a potential oncogene in lung cancer. Exp. Ther. Med. 2017, 13, 3189–3194.
- Sano, O.; Tsujita, M.; Shimizu, Y.; Kato, R.; Kobayashi, A.; Kioka, N.; Remaley, A.T.; Michikawa, M.; Ueda, K.; Matsuo, M. ABCG1 and ABCG4 Suppress gamma-Secretase Activity and Amyloid beta Production. PLoS ONE 2016, 11, e0155400.
- Zhang, Z.; Li, X.; Wang, X.; Zhou, Y.; Xu, H.; Wang, J.; Huang, L.; Tian, Y.; Cheng, Q. The expression of ABCG4, V-ATPase and clinic significance of their correlation with NSCLC. Zhongguo Fei Ai Za Zhi 2008, 11, 691–695.
- Wang, J.; Mitsche, M.A.; Lutjohann, D.; Cohen, J.C.; Xie, X.S.; Hobbs, H.H. Relative roles of ABCG5/ABCG8 in liver and intestine. J. Lipid Res. 2015, 56, 319–330.
- Bastida, J.M.; Benito, R.; Gonzalez-Porras, J.R.; Rivera, J. ABCG5 and ABCG8 gene variations associated with sitosterolemia and platelet dysfunction. Platelets 2021, 32, 573–577.
- Tada, H.; Nohara, A.; Inazu, A.; Sakuma, N.; Mabuchi, H.; Kawashiri, M.A. Sitosterolemia, Hypercholesterolemia, and Coronary Artery Disease. J. Atheroscler. Thromb. 2018, 25, 783–789.
- Wang, H.H.; Liu, M.; Portincasa, P.; Wang, D.Q. Recent Advances in the Critical Role of the Sterol Efflux Transporters ABCG5/G8 in Health and Disease. Adv. Exp. Med. Biol. 2020, 1276, 105–136.
- Chen, Y.N.; Mickley, L.A.; Schwartz, A.M.; Acton, E.M.; Hwang, J.L.; Fojo, A.T. Characterization of adriamycin-resistant human breast cancer cells which display overexpression of a novel resistance-related membrane protein. J. Biol. Chem. 1990, 265, 10073–10080.
- Lee, J.S.; Scala, S.; Matsumoto, Y.; Dickstein, B.; Robey, R.; Zhan, Z.; Altenberg, G.; Bates, S.E. Reduced drug accumulation and multidrug resistance in human breast cancer cells without associated P-glycoprotein or MRP overexpression. J. Cell. Biochem. 1997, 65, 513–526.
- Doyle, L.A.; Yang, W.; Abruzzo, L.V.; Krogmann, T.; Gao, Y.; Rishi, A.K.; Ross, D.D. A multidrug resistance transporter from human MCF-7 breast cancer cells. Proc. Natl. Acad. Sci. USA 1998, 95, 15665–15670.
- Annilo, T.; Chen, Z.Q.; Shulenin, S.; Costantino, J.; Thomas, L.; Lou, H.; Stefanov, S.; Dean, M. Evolution of the vertebrate ABC gene family: Analysis of gene birth and death. Genomics 2006, 88, 1–11.
- Mickley, L.; Jain, P.; Miyake, K.; Schriml, L.M.; Rao, K.; Fojo, T.; Bates, S.; Dean, M. An ATP-binding cassette gene (ABCG3) closely related to the multidrug transporter ABCG2 (MXR/ABCP) has an unusual ATP-binding domain. Mamm. Genome 2001, 12, 86–88.
- Bailey-Dell, K.J.; Hassel, B.; Doyle, L.A.; Ross, D.D. Promoter characterization and genomic organization of the human breast cancer resistance protein (ATP-binding cassette transporter G2) gene. Biochim. Biophys. Acta (BBA)-Gene Struct. Expr. 2001, 1520, 234–241.
- Krishnamurthy, P.; Ross, D.D.; Nakanishi, T.; Bailey-Dell, K.; Zhou, S.; Mercer, K.E.; Sarkadi, B.; Sorrentino, B.P.; Schuetz, J.D. The stem cell marker Bcrp/ABCG2 enhances hypoxic cell survival through interactions with heme. J. Biol. Chem. 2004, 279, 24218–24225.
- Ee, P.L.; Kamalakaran, S.; Tonetti, D.; He, X.; Ross, D.D.; Beck, W.T. Identification of a novel estrogen response element in the breast cancer resistance protein (ABCG2) gene. Cancer Res. 2004, 64, 1247–1251.
- Wang, H.; Lee, E.W.; Zhou, L.; Leung, P.C.; Ross, D.D.; Unadkat, J.D.; Mao, Q. Progesterone receptor (PR) isoforms PRA and PRB differentially regulate expression of the breast cancer resistance protein in human placental choriocarcinoma BeWo cells. Mol. Pharmacol. 2008, 73, 845–854.
- To, K.K.; Robey, R.; Zhan, Z.; Bangiolo, L.; Bates, S.E. Upregulation of ABCG2 by romidepsin via the aryl hydrocarbon receptor pathway. Mol. Cancer Res. 2011, 9, 516–527.
- Szatmari, I.; Vamosi, G.; Brazda, P.; Balint, B.L.; Benko, S.; Szeles, L.; Jeney, V.; Ozvegy-Laczka, C.; Szanto, A.; Barta, E.; et al. Peroxisome proliferator-activated receptor gamma-regulated ABCG2 expression confers cytoprotection to human dendritic cells. J. Biol. Chem. 2006, 281, 23812–23823.
- Honorat, M.; Mesnier, A.; Di Pietro, A.; Lin, V.; Cohen, P.; Dumontet, C.; Payen, L. Dexamethasone down-regulates ABCG2 expression levels in breast cancer cells. Biochem. Biophys. Res. Commun. 2008, 375, 308–314.
- Turner, J.G.; Gump, J.L.; Zhang, C.; Cook, J.M.; Marchion, D.; Hazlehurst, L.; Munster, P.; Schell, M.J.; Dalton, W.S.; Sullivan, D.M. ABCG2 expression, function, and promoter methylation in human multiple myeloma. Blood 2006, 108, 3881–3889.
- Sarkadi, B.; Homolya, L.; Hegedus, T. The ABCG2/BCRP transporter and its variants-from structure to pathology. FEBS Lett. 2020, 594, 4012–4034.
- McDevitt, C.A.; Collins, R.F.; Conway, M.; Modok, S.; Storm, J.; Kerr, I.D.; Ford, R.C.; Callaghan, R. Purification and 3D structural analysis of oligomeric human multidrug transporter ABCG2. Structure 2006, 14, 1623–1632.
- Gyongy, Z.; Mocsar, G.; Hegedus, E.; Stockner, T.; Ritter, Z.; Homolya, L.; Schamberger, A.; Orban, T.I.; Remenyik, J.; Szakacs, G.; et al. Nucleotide binding is the critical regulator of ABCG2 conformational transitions. Elife 2023, 12, e83976.
- Yu, Q.; Ni, D.; Kowal, J.; Manolaridis, I.; Jackson, S.M.; Stahlberg, H.; Locher, K.P. Structures of ABCG2 under turnover conditions reveal a key step in the drug transport mechanism. Nat. Commun. 2021, 12, 4376.
- Jackson, S.M.; Manolaridis, I.; Kowal, J.; Zechner, M.; Taylor, N.M.I.; Bause, M.; Bauer, S.; Bartholomaeus, R.; Bernhardt, G.; Koenig, B.; et al. Structural basis of small-molecule inhibition of human multidrug transporter ABCG2. Nat. Struct. Mol. Biol. 2018, 25, 333–340.
- Robey, R.W.; To, K.K.; Polgar, O.; Dohse, M.; Fetsch, P.; Dean, M.; Bates, S.E. ABCG2: A perspective. Adv. Drug Deliv. Rev. 2009, 61, 3–13.
- Chen, Z.; Shi, T.; Zhang, L.; Zhu, P.; Deng, M.; Huang, C.; Hu, T.; Jiang, L.; Li, J. Mammalian drug efflux transporters of the ATP binding cassette (ABC) family in multidrug resistance: A review of the past decade. Cancer Lett. 2016, 370, 153–164.
- van den Heuvel-Eibrink, M.M.; Wiemer, E.A.; Prins, A.; Meijerink, J.P.; Vossebeld, P.J.; van der Holt, B.; Pieters, R.; Sonneveld, P. Increased expression of the breast cancer resistance protein (BCRP) in relapsed or refractory acute myeloid leukemia (AML). Leukemia 2002, 16, 833–839.
- van den Heuvel-Eibrink, M.M.; van der Holt, B.; Burnett, A.K.; Knauf, W.U.; Fey, M.F.; Verhoef, G.E.; Vellenga, E.; Ossenkoppele, G.J.; Lowenberg, B.; Sonneveld, P. CD34-related coexpression of MDR1 and BCRP indicates a clinically resistant phenotype in patients with acute myeloid leukemia (AML) of older age. Ann. Hematol. 2007, 86, 329–337.
- Steinbach, D.; Sell, W.; Voigt, A.; Hermann, J.; Zintl, F.; Sauerbrey, A. BCRP gene expression is associated with a poor response to remission induction therapy in childhood acute myeloid leukemia. Leukemia 2002, 16, 1443–1447.
- Liu, B.; Li, L.J.; Gong, X.; Zhang, W.; Zhang, H.; Zhao, L. Co-expression of ATP binding cassette transporters is associated with poor prognosis in acute myeloid leukemia. Oncol. Lett. 2018, 15, 6671–6677.
- Bartholomae, S.; Gruhn, B.; Debatin, K.M.; Zimmermann, M.; Creutzig, U.; Reinhardt, D.; Steinbach, D. Coexpression of Multiple ABC-Transporters is Strongly Associated with Treatment Response in Childhood Acute Myeloid Leukemia. Pediatr. Blood Cancer 2016, 63, 242–247.
- Sharom, F.J. ABC multidrug transporters: Structure, function and role in chemoresistance. Pharmacogenomics 2008, 9, 105–127.
- Honjo, Y.; Hrycyna, C.A.; Yan, Q.W.; Medina-Perez, W.Y.; Robey, R.W.; van de Laar, A.; Litman, T.; Dean, M.; Bates, S.E. Acquired mutations in the MXR/BCRP/ABCP gene alter substrate specificity in MXR/BCRP/ABCP-overexpressing cells. Cancer Res. 2001, 61, 6635–6639.
- Ejendal, K.F.; Diop, N.K.; Schweiger, L.C.; Hrycyna, C.A. The nature of amino acid 482 of human ABCG2 affects substrate transport and ATP hydrolysis but not substrate binding. Protein Sci. 2006, 15, 1597–1607.
- Cai, X.; Bikadi, Z.; Ni, Z.; Lee, E.W.; Wang, H.; Rosenberg, M.F.; Mao, Q. Role of basic residues within or near the predicted transmembrane helix 2 of the human breast cancer resistance protein in drug transport. J. Pharmacol. Exp. Ther. 2010, 333, 670–681.
- Miwa, M.; Tsukahara, S.; Ishikawa, E.; Asada, S.; Imai, Y.; Sugimoto, Y. Single amino acid substitutions in the transmembrane domains of breast cancer resistance protein (BCRP) alter cross resistance patterns in transfectants. Int. J. Cancer 2003, 107, 757–763.
- Wakabayashi, K.; Nakagawa, H.; Tamura, A.; Koshiba, S.; Hoshijima, K.; Komada, M.; Ishikawa, T. Intramolecular disulfide bond is a critical check point determining degradative fates of ATP-binding cassette (ABC) transporter ABCG2 protein. J. Biol. Chem. 2007, 282, 27841–27846.
- Li, R.; Miao, L.; Qin, L.; Xiang, Y.; Zhang, X.; Peng, H.; Mailamuguli; Sun, Y.; Yao, H. A meta-analysis of the associations between the Q141K and Q126X ABCG2 gene variants and gout risk. Int. J. Clin. Exp. Pathol. 2015, 8, 9812–9823.
- Zhang, L.; Spencer, K.L.; Voruganti, V.S.; Jorgensen, N.W.; Fornage, M.; Best, L.G.; Brown-Gentry, K.D.; Cole, S.A.; Crawford, D.C.; Deelman, E.; et al. Association of functional polymorphism rs2231142 (Q141K) in the ABCG2 gene with serum uric acid and gout in 4 US populations: The PAGE Study. Am. J. Epidemiol. 2013, 177, 923–932.
- Jabalee, J.; Towle, R.; Garnis, C. The Role of Extracellular Vesicles in Cancer: Cargo, Function, and Therapeutic Implications. Cells 2018, 7, 93.
- Safaei, R.; Larson, B.J.; Cheng, T.C.; Gibson, M.A.; Otani, S.; Naerdemann, W.; Howell, S.B. Abnormal lysosomal trafficking and enhanced exosomal export of cisplatin in drug-resistant human ovarian carcinoma cells. Mol. Cancer Ther. 2005, 4, 1595–1604.
- Goler-Baron, V.; Assaraf, Y.G. Structure and function of ABCG2-rich extracellular vesicles mediating multidrug resistance. PLoS ONE 2011, 6, e16007.
- Samuel, P.; Fabbri, M.; Carter, D.R.F. Mechanisms of Drug Resistance in Cancer: The Role of Extracellular Vesicles. Proteomics 2017, 17, 23–24.
- Distefano, M.; Scambia, G.; Ferlini, C.; Gaggini, C.; De Vincenzo, R.; Riva, A.; Bombardelli, E.; Ojima, I.; Fattorossi, A.; Panici, P.B.; et al. Anti-proliferative activity of a new class of taxanes (14beta-hydroxy-10-deacetylbaccatin III derivatives) on multidrug-resistance-positive human cancer cells. Int. J. Cancer 1997, 72, 844–850.
- Shionoya, M.; Jimbo, T.; Kitagawa, M.; Soga, T.; Tohgo, A. DJ-927, a novel oral taxane, overcomes P-glycoprotein-mediated multidrug resistance in vitro and in vivo. Cancer Sci. 2003, 94, 459–466.
- Glavinas, H.; Krajcsi, P.; Cserepes, J.; Sarkadi, B. The role of ABC transporters in drug resistance, metabolism and toxicity. Curr. Drug Deliv. 2004, 1, 27–42.
- Szakacs, G.; Hall, M.D.; Gottesman, M.M.; Boumendjel, A.; Kachadourian, R.; Day, B.J.; Baubichon-Cortay, H.; Di Pietro, A. Targeting the Achilles heel of multidrug-resistant cancer by exploiting the fitness cost of resistance. Chem. Rev. 2014, 114, 5753–5774.
- Rabindran, S.K.; He, H.; Singh, M.; Brown, E.; Collins, K.I.; Annable, T.; Greenberger, L.M. Reversal of a novel multidrug resistance mechanism in human colon carcinoma cells by fumitremorgin C. Cancer Res. 1998, 58, 5850–5858.
- Guragossian, N.; Belhani, B.; Moreno, A.; Nunes, M.T.; Gonzalez-Lobato, L.; Marminon, C.; Berthier, L.; Rocio Andrade Pires, A.D.; Ozvegy-Laczka, C.; Sarkadi, B.; et al. Uncompetitive nanomolar dimeric indenoindole inhibitors of the human breast cancer resistance pump ABCG2. Eur. J. Med. Chem. 2021, 211, 113017.
- Yang, D.; Kathawala, R.J.; Chufan, E.E.; Patel, A.; Ambudkar, S.V.; Chen, Z.S.; Chen, X. Tivozanib reverses multidrug resistance mediated by ABCB1 (P-glycoprotein) and ABCG2 (BCRP). Future Oncol. 2014, 10, 1827–1841.
- Sen, R.; Natarajan, K.; Bhullar, J.; Shukla, S.; Fang, H.B.; Cai, L.; Chen, Z.S.; Ambudkar, S.V.; Baer, M.R. The novel BCR-ABL and FLT3 inhibitor ponatinib is a potent inhibitor of the MDR-associated ATP-binding cassette transporter ABCG2. Mol. Cancer Ther. 2012, 11, 2033–2044.
- Elsby, R.; Martin, P.; Surry, D.; Sharma, P.; Fenner, K. Solitary Inhibition of the Breast Cancer Resistance Protein Efflux Transporter Results in a Clinically Significant Drug-Drug Interaction with Rosuvastatin by Causing up to a 2-Fold Increase in Statin Exposure. Drug Metab. Dispos. 2016, 44, 398–408.
- Miyata, H.; Takada, T.; Toyoda, Y.; Matsuo, H.; Ichida, K.; Suzuki, H. Identification of Febuxostat as a New Strong ABCG2 Inhibitor: Potential Applications and Risks in Clinical Situations. Front. Pharmacol. 2016, 7, 518.
- Sorf, A.; Sucha, S.; Morell, A.; Novotna, E.; Staud, F.; Zavrelova, A.; Visek, B.; Wsol, V.; Ceckova, M. Targeting Pharmacokinetic Drug Resistance in Acute Myeloid Leukemia Cells with CDK4/6 Inhibitors. Cancers 2020, 12, 1596.
- Sabet, Z.; Vagiannis, D.; Budagaga, Y.; Zhang, Y.; Novotna, E.; Hanke, I.; Rozkos, T.; Hofman, J. Talazoparib Does Not Interact with ABCB1 Transporter or Cytochrome P450s, but Modulates Multidrug Resistance Mediated by ABCC1 and ABCG2: An in Vitro and Ex Vivo Study. Int. J. Mol. Sci. 2022, 23, 14338.
- Damiani, D.; Tiribelli, M.; Geromin, A.; Cerno, M.; Zanini, F.; Michelutti, A.; Fanin, R. ABCG2, Cytogenetics, and Age Predict Relapse after Allogeneic Stem Cell Transplantation for Acute Myeloid Leukemia in Complete Remission. Biol. Blood Marrow Transpl. 2016, 22, 1621–1626.
This entry is offline, you can click here to edit this entry!