Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Mutations in the genes that encode for lamins, predominantly lamin A/C, cause a wide spectrum of human diseases, referred to as laminopathies, including muscular dystrophy, lipodystrophy, and systemic premature aging syndrome. HGPS, one of the most severe laminopathies, is a rare genetic disorder characterized by multisystem abnormalities, including premature aging. The most frequent mutation causing HGPS is c.1842C>T (p.G608G) in exon 11 of LMNA, resulting in cryptic splicing between an abnormal donor site in the middle of exon 11 and the usual acceptor of exon 12. This change causes a 50-amino acid deletion in the carboxyl-terminal tail of prelamin A, producing a truncated protein referred to as progerin. The 50 missing amino acids include the recognition sites for the prelamin A-cleaving enzyme ZMPSTE24. Consequently, progerin is normally farnesylated but cannot be further processed because of the lack of docking sites for ZMPSTE24. The farnesylated domain of progerin is firmly anchored to the nuclear membrane, leading to nuclear deformation and deleterious effects in HGPS cells. Blocking the farnesylation of progerin with a farnesyltransferase inhibitor (FTI) successfully reduced the cytotoxic effects of progerin in vitro; however, a clinical trial of FTIs did not yield promising results. A study showed that the interaction between progerin and wild-type lamin A/C was also a critical cause of nuclear deformation in HGPS and normal aging cells, providing a new therapeutic target for HGPS. Progerin expression in various cell types induces excessive ROS production and reduces the activities of the antioxidant system. Oxidative stress is also implicated in other types of laminopathies, such as Dunnigan-type familial partial lipodystrophy (FPLD), amyotrophic quadricipital syndrome with cardiac involvement, autosomal dominant Emery–Dreifuss muscular dystrophy (AD-EDMD), and restrictive dermopathy (RD).
- lamins
- laminopathy
- HGPS
- oxidative stress
1. Effects of Excessive ROS
The downstream effects of oxidative stress, particularly those induced by progerin or other types of mutant lamin A, are characterized by persistent DNA damage and telomere shortening [1][2][3]. Fibroblasts from HGPS patients have shown higher ROS levels than those from age-matched controls [4]. Many studies have shown that fibroblasts with HGPS exhibit persistent H2AX foci [5], increased phospho-H2AX foci [6], altered H3 methylation, and HP1 downregulation, which are indicative of the accumulation of unrepaired DNA damage [7][8][9][10]. Zmpste24−/− mouse embryonic fibroblasts (MEFs) accumulate farnesylated prelamin A, exhibit increased DNA damage, and are more sensitive to DNA-damaging agents [5]. Bone marrow cells from Zmpste24-deficient mice show increased aneuploidy. Zmpste24−/− MEFs and HGPS fibroblasts display impaired DNA damage responses characterized by delayed recruitment of DNA damage proteins Rad50 and 53BP1 (p53-binding protein 1) to the damage sites. The presence of progerin also triggers the loss of components of the NuRD complex, which contributes to persistent DNA damage and genomic instability [6]. Interestingly, the basal levels of double-strand breaks (DSBs) in HGPS fibroblasts were reduced upon treatment with ROS scavengers, such as N-acetyl cysteine (NAC) [7] and sulforaphane [11]. Additionally, treatment with antioxidants, such as all-trans retinoic acid (ATRA), rescued cellular dynamics and proliferation in HGPS fibroblasts via recovery of the DNA damage response factor PARP1 [12]. Together, these results indicate that the interplay between mutant lamin A and oxidative stress leads to persistent cellular DNA damage.
In most somatic cells, telomere shortening is caused by decreased telomerase activity. It has been proposed that oxidative stress causes telomere shortening. In a study of fibroblasts isolated from humans and sheep, ROS accumulation and the telomere shortening rate showed a continuous, exponential correlation, regardless of the species [3]. It has also been shown that telomerases are exported from the nucleus to the cytosol upon excessive ROS accumulation [13]. Telomere shortening causes ROS accumulation, and it is well known that telomere shortening activates the p53 tumor suppressor to progressively induce cell cycle arrest, senescence, and apoptosis in response to genotoxic stimuli. Activation of p53 represses PGC-1α and PGC-1β genes on their promoters [14]. Since PGC-1α and PGC-1β are master regulators of mitochondrial physiology and metabolism, telomere shortening causes defects in mitochondrial biogenesis and function and the consequent accumulation of ROS. Furthermore, the telomere shortening rate correlates with the accumulation of DNA damage [15]. Overexpression of mutant LMNA variants in human fibroblasts led to accelerated telomere shortening and cellular senescence [16]. In the study, the overexpression of wild-type LMNA also resulted in increased telomere shortening. Oxidative stress also affects the lamin structure. Conserved cysteine residues in mammalian lamins A/C are required for cellular defense against ROS [17]. ROS can oxidize these residues and perturb their function, rendering them vulnerable to ROS-mediated damage. Therefore, it is plausible that changes in the nuclear architecture, due to the expression of either mutant or wild-type LMNA, initiate a reinforcing loop consisting of telomere shortening, ROS accumulation, and DNA damage.
2. Generation of Excessive ROS by Alterations in Lamin A/C
Mitochondrial dysfunction is a major source of ROS in laminopathies. Sustained accumulation of farnesylated prelamin A or depletion of normal lamin A/C results in mitochondrial dysfunction and oxidative stress in human fibroblasts [18]. The expression of cytochrome c oxidase subunit II (COX 2) is drastically decreased in human fibroblasts and adipocytes bearing lamin A variants, which lowers mitochondrial membrane potential and generates excessive ROS [19]. Lmna-null MEFs and human HGPS fibroblasts exhibit reduced NAD+ levels, unstable mitochondrial DNA, and weakened bioenergetics [20]. This dysfunction is linked to reduced PGC-1α levels, and diminished expression of NAD+ biosynthesis enzyme NAMPT and NAD+-dependent deacetylase SIRT1. It also showed that lamin A/C aberrations lead to high levels of PARylation, which further lowers the NAD+ pool and contributes to impaired DNA base excision repair under oxidative stress. Impaired nuclear transport is another source of excessive ROS production in laminopathies. A study showed that the altered sensitivity of Ran to large cargo transport across the nuclear membrane contributed to disease-associated phenotypes in HGPS fibroblasts [21]. Another study found that in Drosophila, certain transcripts, such as those encoding the mitochondrial assembly regulatory factor (Marf) and a mitochondrial fusion factor (mitofusin), exit the nucleus through NE budding [22]. However, abnormal lamina organization caused by lamin C mutation inhibits the release of these RNAs via NE budding, leading to impaired RNA export and progressive deterioration of mitochondrial integrity, ultimately resulting in premature aging.
3. Defects in the Antioxidant System in Laminopathies
The expression levels of primary antioxidant enzymes are altered in laminopathies. Catalase (CAT) and glutathione peroxidase (GPX) activities in HGPS fibroblasts were only 50% and 30%, respectively, compared to those in normal fibroblasts [23]. However, other studies have reported elevated levels of superoxide dismutase (SOD), CAT, and glutathione (GSH)-S-transferase (GST) in fibroblasts isolated from patients with LmnaY259X/Y259X [17][24].
Peroxisomes are subcellular organelles surrounded by membranes in which many catalytic enzymes play critical roles in scavenging ROS. Proteins targeting peroxisomes were found to be defective in fibroblasts isolated from patients with HGPS [25]. In the study, aberrant peroxisomes referred to as “peroxisome ghosts”, which lacked some of the matrix proteins, were abundant in HGPS fibroblasts. In addition, CAT activity was reduced, and consequently, high levels of ROS were observed in fibroblasts expressing progerin. Therefore, the presence of progerin sequentially leads to impaired peroxisome protein targeting, defective peroxisome assembly, reduced activity of ROS-defusing enzymes, particularly CAT, and high levels of ROS, which contribute to premature senescence phenotypes. Mature erythrocytes are constantly exposed to oxidative stress more frequently than other cell types and therefore have a highly efficient antioxidant system, particularly related to GSH. In erythrocytes, reduced GSH expression plays a prominent role in mitigating the consequent damage [26]. In a rat model of HGPS, erythrocytes showed impaired membrane integrity and oxidation of transport proteins in the presence of progerin, which decreased L-cysteine influx [27]. The decreased L-cysteine influx was attributed to the decreased erythrocyte GSH content observed in HGPS rats.
4. The Emerging Role of the Nrf2 Pathway in HGPS and Normal Aging
Nuclear factor erythroid 2-related factor 2 (Nrf2) is the master transcription factor for cytoprotective genes against oxidative stress [28]. Under normal conditions, Nrf2 is recognized by Keap1, the substrate recognition subunit (SRS) of Cul3Keap1 E3 ubiquitin ligase, is ubiquitinated, and is finally degraded by the 26S proteasome. However, when cells are exposed to oxidative stress, Keap1 is inactivated and Nrf2 is simultaneously released from the Cul3Keap1 E3 ubiquitin ligase (Figure 1). Free Nrf2 translocates to the nucleus, where it binds to the antioxidant response elements (AREs) of genes involved in antioxidant defense and detoxification pathways and activates their expression. This activation process helps alleviate cellular damage caused by oxidative stress and sustains cellular health. The downstream targets of Nrf2 include genes involved in GSH production and regulation, ROS and xenobiotic detoxification, thioredoxin-based antioxidant systems, NADPH regeneration, and heme and ion metabolism.
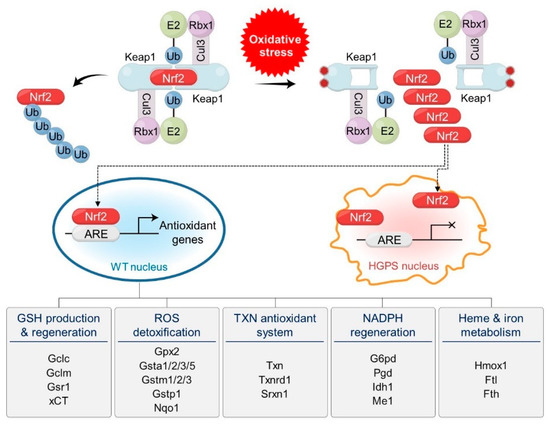
Figure 1. Oxidative stress and progerin are implicated in the regulation of Nrf2, the master transcription factor for cellular defense systems against oxidative stress. Dimeric Cul3Keap1 E3 ubiquitin ligase complexes target Nrf2 for polyubiquitination and degradation under normal conditions. Under oxidative stress, cysteine residues of Keap1 are oxidized and the structure of Nrf2-binding motifs is modified, which releases Nrf2 from Cul3 ligase complexes. Released Nrf2 translocates into the nucleus and activates antioxidant genes by binding to antioxidant response elements (AREs) in normal cells. Over 200 antioxidant genes are activated by Nrf2. They are classified by their functions and include genes involved in GSH production and regeneration, ROS detoxification, TXN antioxidant systems, NADPH regeneration, and heme and iron metabolism. Representative genes are indicated under the functions. In HGPS cells, progerin sequesters Nrf2 at the nuclear periphery, which prevents the activation of antioxidant genes.
High-throughput screening identified CAND1, a negative regulator of Nrf2, as an effector of aging defects in HGPS [29]. The study showed that Nrf2 transcriptional activity was impaired in HGPS, and reactivation of Nrf2 reversed these defects in HGPS fibroblasts. In contrast, Nrf2 knockdown induced oxidative stress and HGPS-like defects in wild-type fibroblasts. Therefore, the downregulation of the Nrf2 pathway and consequent elevation of ROS levels may be the key mechanisms that induce various defects in HGPS. An investigation into the molecular mechanism showed that progerin has a 2–3-fold higher affinity for Nrf2 compared to wild-type lamin A in both in vivo and in vitro assays. Microscopic observation revealed that Nrf2 colocalized with progerin but not with wild-type lamin A at the nuclear periphery, suggesting its sequestration by progerin. Therefore, it has been postulated that progerin-sequestered Nrf2, particularly at the nuclear periphery, cannot bind to ARS motifs in its target genes, which might impair the cellular defense system against oxidative stress in HGPS and during normal aging.
In addition, wild-type lamin A interacts with sirtuin 6 (SIRT6), which is implicated in multiple molecular pathways, including DNA repair, telomere maintenance, and anti-inflammatory pathways [30]. Wild-type lamin A, but not progerin, enhances the deacetylase activity and efficient chromatin localization of SIRT6 to promote its functions, including DNA repair. Consistent with this finding, SIRT6 activity is defective in HGPS fibroblasts. Therefore, in the presence of progerin, wild-type lamin A may not efficiently activate or localize SIRT6 to DNA lesions in the chromatin. Moreover, several studies have shown that SIRT6 promotes the Nrf2 target gene expression [31][32]. A recent study found a strong association between progerin and ER stress in the etiology of HGPS [33]. It has been also shown that alteration in the NL causes NF-κB activation and induces proinflammatory cytokines [34]. Nrf2 plays a crucial role in ER stress and NF-κB pathways [35]. Progerin and Nrf2 are interconnected and impair the formation of functional complexes that promote various cellular defense systems against oxidative stress.
This entry is adapted from the peer-reviewed paper 10.3390/cells12091234
References
- Decker, M.L.; Chavez, E.; Vulto, I.; Lansdorp, P.M. Telomere length in Hutchinson-Gilford progeria syndrome. Mech. Ageing Dev. 2009, 130, 377–383.
- Hutchison, C.J. The role of DNA damage in laminopathy progeroid syndromes. Biochem. Soc. Trans. 2011, 39, 1715–1718.
- Richter, T.; Zglinicki, T.v. A continuous correlation between oxidative stress and telomere shortening in fibroblasts. Exp. Gerontol. 2007, 42, 1039–1042.
- Viteri, G.; Chung, Y.W.; Stadtman, E.R. Effect of progerin on the accumulation of oxidized proteins in fibroblasts from Hutchinson Gilford progeria patients. Mech. Ageing Dev. 2010, 131, 2–8.
- Liu, B.; Wang, J.; Chan, K.M.; Tjia, W.M.; Deng, W.; Guan, X.; Huang, J.D.; Li, K.M.; Chau, P.Y.; Chen, D.J.; et al. Genomic instability in laminopathy-based premature aging. Nat. Med. 2005, 11, 780–785.
- Pegoraro, G.; Kubben, N.; Wickert, U.; Göhler, H.; Hoffmann, K.; Misteli, T. Ageing-related chromatin defects through loss of the NURD complex. Nat. Cell Biol. 2009, 11, 1261–1267.
- Richards, S.A.; Muter, J.; Ritchie, P.; Lattanzi, G.; Hutchison, C.J. The accumulation of un-repairable DNA damage in laminopathy progeria fibroblasts is caused by ROS generation and is prevented by treatment with N-acetyl cysteine. Hum. Mol. Genet. 2011, 20, 3997–4004.
- Scaffidi, P.; Misteli, T. Lamin A-dependent nuclear defects in human aging. Science 2006, 312, 1059–1063.
- di Masi, A.; D’Apice, M.R.; Ricordy, R.; Tanzarella, C.; Novelli, G. The R527H mutation in LMNA gene causes an increased sensitivity to ionizing radiation. Cell Cycle 2008, 7, 2030–2037.
- Shumaker, D.K.; Dechat, T.; Kohlmaier, A.; Adam, S.A.; Bozovsky, M.R.; Erdos, M.R.; Eriksson, M.; Goldman, A.E.; Khuon, S.; Collins, F.S.; et al. Mutant nuclear lamin A leads to progressive alterations of epigenetic control in premature aging. Proc. Natl. Acad. Sci. USA 2006, 103, 8703–8708.
- Gabriel, D.; Roedl, D.; Gordon, L.B.; Djabali, K. Sulforaphane enhances progerin clearance in Hutchinson–Gilford progeria fibroblasts. Aging Cell 2015, 14, 78–91.
- Pellegrini, C.; Columbaro, M.; Capanni, C.; D’Apice, M.R.; Cavallo, C.; Murdocca, M.; Lattanzi, G.; Squarzoni, S. All-trans retinoic acid and rapamycin normalize Hutchinson Gilford progeria fibroblast phenotype. Oncotarget 2015, 6, 29914–29928.
- Haendeler, J.; Hoffmann, J.; Brandes, R.P.; Zeiher, A.M.; Dimmeler, S. Hydrogen peroxide triggers nuclear export of telomerase reverse transcriptase via Src kinase family-dependent phosphorylation of tyrosine 707. Mol. Cell Biol. 2003, 23, 4598–4610.
- Sahin, E.; Colla, S.; Liesa, M.; Moslehi, J.; Muller, F.L.; Guo, M.; Cooper, M.; Kotton, D.; Fabian, A.J.; Walkey, C.; et al. Telomere dysfunction induces metabolic and mitochondrial compromise. Nature 2011, 470, 359–365.
- He, S.; Sharpless, N.E. Senescence in Health and Disease. Cell 2017, 169, 1000–1011.
- Huang, S.; Risques, R.A.; Martin, G.M.; Rabinovitch, P.S.; Oshima, J. Accelerated telomere shortening and replicative senescence in human fibroblasts overexpressing mutant and wild-type lamin A. Exp. Cell Res. 2008, 314, 82–91.
- Pekovic, V.; Gibbs-Seymour, I.; Markiewicz, E.; Alzoghaibi, F.; Benham, A.M.; Edwards, R.; Wenhert, M.; von Zglinicki, T.; Hutchison, C.J. Conserved cysteine residues in the mammalian lamin A tail are essential for cellular responses to ROS generation. Aging Cell 2011, 10, 1067–1079.
- Sieprath, T.; Corne, T.D.; Nooteboom, M.; Grootaert, C.; Rajkovic, A.; Buysschaert, B.; Robijns, J.; Broers, J.L.; Ramaekers, F.C.; Koopman, W.J.; et al. Sustained accumulation of prelamin A and depletion of lamin A/C both cause oxidative stress and mitochondrial dysfunction but induce different cell fates. Nucleus 2015, 6, 236–246.
- Caron, M.; Auclair, M.; Donadille, B.; Béréziat, V.; Guerci, B.; Laville, M.; Narbonne, H.; Bodemer, C.; Lascols, O.; Capeau, J.; et al. Human lipodystrophies linked to mutations in A-type lamins and to HIV protease inhibitor therapy are both associated with prelamin A accumulation, oxidative stress and premature cellular senescence. Cell Death Differ. 2007, 14, 1759–1767.
- Maynard, S.; Hall, A.; Galanos, P.; Rizza, S.; Yamamoto, T.; Gram, H.H.; Munk, S.H.N.; Shoaib, M.; Sorensen, C.S.; Bohr, V.A.; et al. Lamin A/C impairments cause mitochondrial dysfunction by attenuating PGC1alpha and the NAMPT-NAD+ pathway. Nucleic Acids Res. 2022, 50, 9948–9965.
- Snow, C.J.; Dar, A.; Dutta, A.; Kehlenbach, R.H.; Paschal, B.M. Defective nuclear import of Tpr in Progeria reflects the Ran sensitivity of large cargo transport. J. Cell Biol. 2013, 201, 541–557.
- Li, Y.; Hassinger, L.; Thomson, T.; Ding, B.; Ashley, J.; Hassinger, W.; Budnik, V. Lamin Mutations Accelerate Aging via Defective Export of Mitochondrial mRNAs through Nuclear Envelope Budding. Curr. Biol. 2016, 26, 2052–2059.
- Yan, T.; Li, S.; Jiang, X.; Oberley, L.W. Altered levels of primary antioxidant enzymes in progeria skin fibroblasts. Biochem Biophys. Res. Commun. 1999, 257, 163–167.
- De Vos, W.H.; Houben, F.; Kamps, M.; Malhas, A.; Verheyen, F.; Cox, J.; Manders, E.M.; Verstraeten, V.L.; van Steensel, M.A.; Marcelis, C.L.; et al. Repetitive disruptions of the nuclear envelope invoke temporary loss of cellular compartmentalization in laminopathies. Hum. Mol. Genet. 2011, 20, 4175–4186.
- Mao, X.; Bharti, P.; Thaivalappil, A.; Cao, K. Peroxisomal abnormalities and catalase deficiency in Hutchinson-Gilford Progeria Syndrome. Aging 2020, 12, 5195–5208.
- Gwozdzinski, K.; Pieniazek, A.; Tabaczar, S.; Jegier, A.; Brzeszczynska, J. Investigation of oxidative stress parameters in different lifespan erythrocyte fractions in young untrained men after acute exercise. Exp. Physiol. 2017, 102, 190–201.
- Chaudhary, M.K.; Rizvi, S.I. Erythrocyte Senescence in a Model of Rat Displaying Hutchinson-Gilford Progeria Syndrome. Anal. Cell. Pathol. 2018, 2018, 5028925.
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional Regulation by Nrf2. Antioxid. Redox Signal. 2018, 29, 1727–1745.
- Kubben, N.; Zhang, W.; Wang, L.; Voss, T.C.; Yang, J.; Qu, J.; Liu, G.-H.; Misteli, T. Repression of the Antioxidant NRF2 Pathway in Premature Aging. Cell 2016, 165, 1361–1374.
- Ghosh, S.; Liu, B.; Wang, Y.; Hao, Q.; Zhou, Z. Lamin A Is an Endogenous SIRT6 Activator and Promotes SIRT6-Mediated DNA Repair. Cell Rep. 2015, 13, 1396–1406.
- Liu, X.; Ren, S.; Li, Z.; Hao, D.; Zhao, X.; Zhang, Z.; Liu, D. Sirt6 mediates antioxidative functions by increasing Nrf2 abundance. Exp. Cell Res. 2023, 422, 113409.
- Rezazadeh, S.; Yang, D.; Tombline, G.; Simon, M.; Regan, S.P.; Seluanov, A.; Gorbunova, V. SIRT6 promotes transcription of a subset of NRF2 targets by mono-ADP-ribosylating BAF170. Nucleic Acids Res. 2019, 47, 7914–7928.
- Hamczyk, M.R.; Villa-Bellosta, R.; Quesada, V.; Gonzalo, P.; Vidak, S.; Nevado, R.M.; Andres-Manzano, M.J.; Misteli, T.; Lopez-Otin, C.; Andres, V. Progerin accelerates atherosclerosis by inducing endoplasmic reticulum stress in vascular smooth muscle cells. EMBO Mol. Med. 2019, 11, e9736.
- Osorio, F.G.; Barcena, C.; Soria-Valles, C.; Ramsay, A.J.; de Carlos, F.; Cobo, J.; Fueyo, A.; Freije, J.M.; Lopez-Otin, C. Nuclear lamina defects cause ATM-dependent NF-kappaB activation and link accelerated aging to a systemic inflammatory response. Genes Dev. 2012, 26, 2311–2324.
- Mozzini, C.; Setti, A.; Cicco, S.; Pagani, M. The Most Severe Paradigm of Early Cardiovascular Disease: Hutchinson-Gilford Progeria. Focus on the Role of Oxidative Stress. Curr. Probl. Cardiol. 2022, 47, 100900.
This entry is offline, you can click here to edit this entry!