Parkinson’s disease (PD) is an age-related chronic, progressive, multi-system, neurodegenerative disease with an incidence second only to Alzheimer’s disease. A PD diagnosis requires the presence of two core motor features, including diminished movement (bradykinesia), tremor, muscle rigidity, or postural instability, difficulty initiating voluntary movement (akinesia), involuntary eye movements, and blinking, which can take up to 15-20 years to become evident. There is now an increasing level of evidence linking mitochondrial dysfunction, including elevated oxidative stress, reactive oxygen species (ROS) and impaired cellular energy production, with the overactivation and escalation of a microglial mediated proinflammatory immune response, as naturally occurring and damaging interlinked bidirectional and self-perpetuating cycles that share common pathological processes in aging and PD. This paper proposes that both chronic inflammation, microglial activation and neuronal mitochondrial impairment should be considered as concurrently influencing each other along a continuum, rather than as separate and isolated linear metabolic events that affect specific aspects of neural processing and brain function.
- alpha-synuclein
- adenosine triphosphate
- bioenergetic capacity
- cytokines
- dopamine neurons
1. Age as the Key Risk Factor of PD
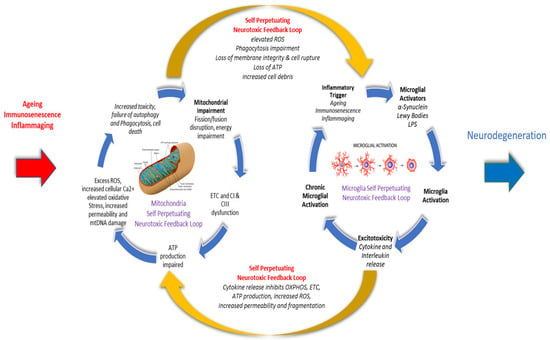
2. Inflammation and Inflammaging in Ageing
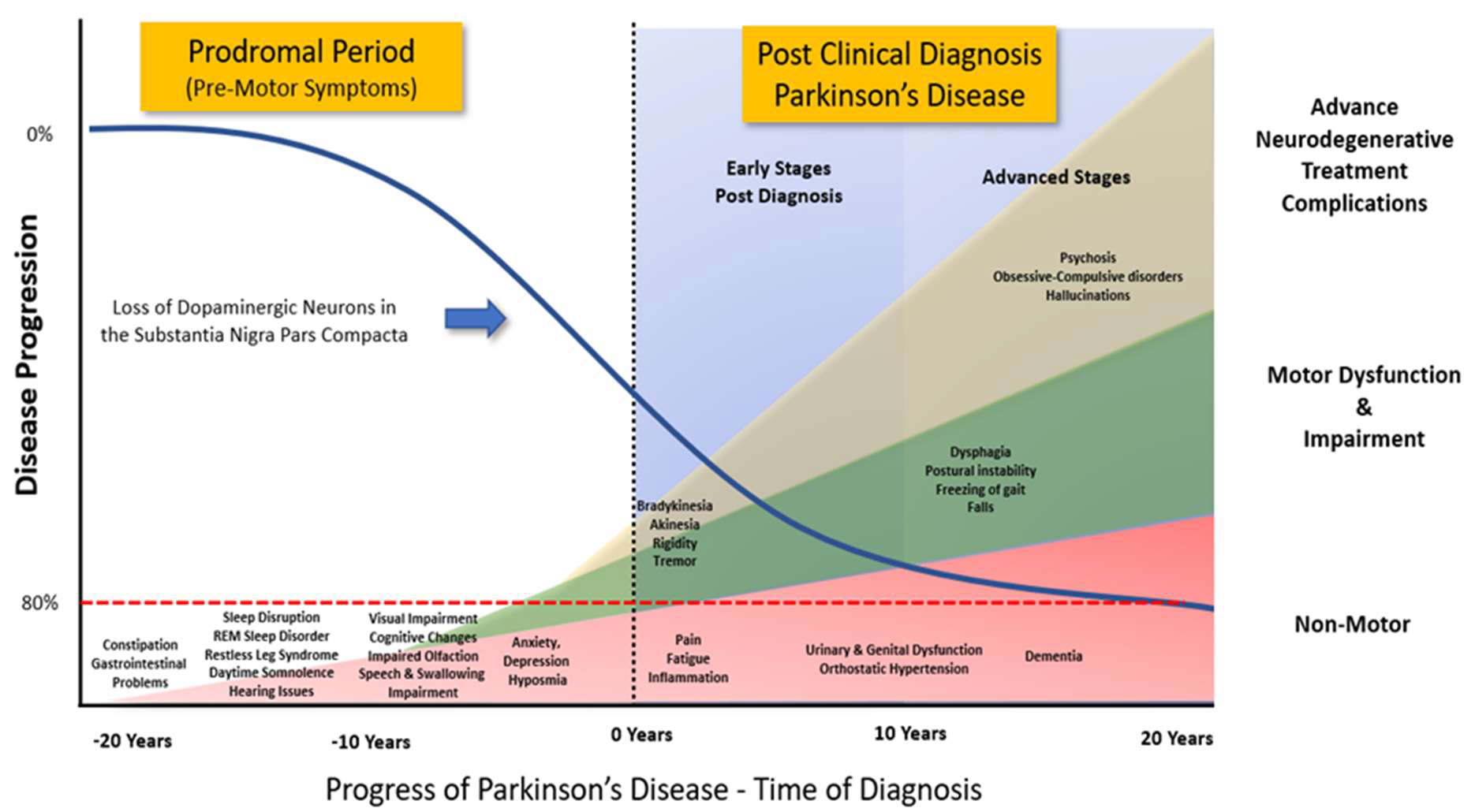
Figure 2. Progression of Clinical Symptoms from Early Prodromal through to Clinical Diagnosis of Parkinson’s Disease. Clinical diagnosis of Parkinson’s Disease accepts that there is a potential loss of up to 80% of the dopaminergic neurons in the Substantia Nigra Pars Compacta. However, prior to the emergence of any significant motor impairment, a wide variety of symptoms associated with non-motor dysfunction and disability usually precede the clinical diagnosis of Parkinson’s disease by 10-20 years. (Adapted from data and figures in Kalia et al., [61], Parkinson's disease. The Lancet, 386(9996), 896-912, and Tansey et al., [62], Inflammation and immune dysfunction in Parkinson disease.
3. Mitochondrial Dysfunction and PD
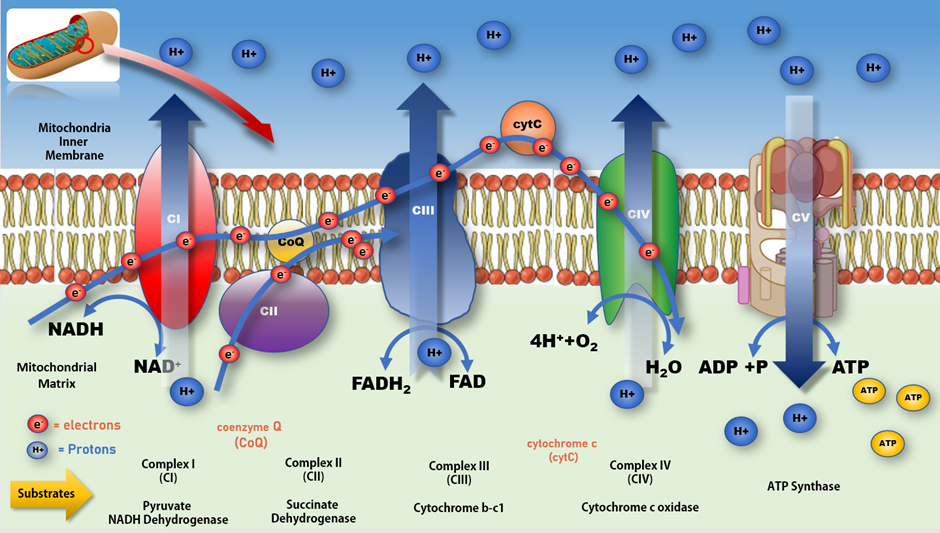
Figure 3. Mitochondrial electron transport chain and production of ATP. Impeding the flow of electrons along the ETC at CI increases production of free radicals resulting in a rise in oxidative stress and lower production of ATP at CV. A more complete review of mitochondrial bioenergetic function and associated impacts on various disease processes can be found in Protasoni et al. 2021 [70].
This entry is adapted from the peer-reviewed paper 10.3390/antiox12051117
References
- Azam, S.; Haque, M.E.; Kim, I.S.; Choi, D.K. Microglial Turnover in Ageing-Related Neurodegeneration: Therapeutic Avenue to Intervene in Disease Progression. Cells 2021, 10, 150.
- Oberg, M.; Fabrik, I.; Fabrikova, D.; Zehetner, N.; Hartlova, A. The role of innate immunity and inflammation in Parkinson s disease. Scand. J. Immunol. 2021, 93, e13022.
- Currais, A.; Fischer, W.; Maher, P.; Schubert, D. Intraneuronal protein aggregation as a trigger for inflammation and neurodegeneration in the aging brain. FASEB J. 2017, 31, 5–10.
- Currais, A. Ageing and inflammation—A central role for mitochondria in brain health and disease. Ageing Res. Rev. 2015, 21, 30–42.
- Gonzalez, H.; Elgueta, D.; Montoya, A.; Pacheco, R. Neuroimmune regulation of microglial activity involved in neuroinflammation and neurodegenerative diseases. J. Neuroimmunol. 2014, 274, 1–13.
- Ren, M.; Guo, Y.; Wei, X.; Yan, S.; Qin, Y.; Zhang, X.; Jiang, F.; Lou, H. TREM2 overexpression attenuates neuroinflammation and protects dopaminergic neurons in experimental models of Parkinson’s disease. Exp. Neurol. 2018, 302, 205–213.
- Tansey, M.G.; Goldberg, M.S. Neuroinflammation in Parkinson’s disease: Its role in neuronal death and implications for therapeutic intervention. Neurobiol. Dis. 2010, 37, 510–518.
- Subhramanyam, C.S.; Wang, C.; Hu, Q.; Dheen, S.T. Microglia-mediated neuroinflammation in neurodegenerative diseases. Semin. Cell Dev. Biol. 2019, 94, 112–120.
- Schain, M.; Kreisl, W.C. Neuroinflammation in Neurodegenerative Disorders-a Review. Curr. Neurol. Neurosci. Rep. 2017, 17, 25.
- Rango, M.; Bresolin, N. Brain Mitochondria, Aging, and Parkinson’s Disease. Genes 2018, 9, 250.
- McGuire, P.J. Mitochondrial Dysfunction and the Aging Immune System. Biology 2019, 8, 26.
- Lenaz, G.; D’Aurelio, M.; Merlo Pich, M.; Genova, M.L.; Ventura, B.; Bovina, C.; Formiggini, G.; Parenti Castelli, G. Mitochondrial bioenergetics in aging. Biochim. Biophys. Acta (BBA) Bioenerg. 2000, 1459, 397–404.
- Chandra, G.; Shenoi, R.; Anand, R.; Rajamma, U.; Mohanakumar, K. Reinforcing mitochondrial functions in aging brain: An insight into Parkinson’s disease therapeutics. J. Chem. Neuroanat. 2019, 95, 29–42.
- Pence, B.D.; Yarbro, J.R. Aging impairs mitochondrial respiratory capacity in classical monocytes. Exp. Gerontol. 2018, 108, 112–117.
- Koziorowski, D.; Figura, M.; Milanowski, Ł.M.; Szlufik, S.; Alster, P.; Madetko, N.; Friedman, A. Mechanisms of Neurodegeneration in Various Forms of Parkinsonism, Similarities and Differences. Cells 2021, 10, 656.
- Giummarra, L.; Crewther, S.G.; Riddell, N.; Murphy, M.J.; Crewther, D.P. Pathway analysis identifies altered mitochondrial metabolism, neurotransmission, structural pathways and complement cascade in retina/RPE/choroid in chick model of form-deprivation myopia. PeerJ 2018, 6, e5048.
- Culmsee, C.; Michels, S.; Scheu, S.; Arolt, V.; Dannlowski, U.; Alferink, J. Mitochondria, Microglia, and the Immune System-How Are They Linked in Affective Disorders? Front. Psychiatry 2018, 9, 739.
- Van Horssen, J.; van Schaik, P.; Witte, M. Inflammation and mitochondrial dysfunction: A vicious circle in neurodegenerative disorders? Neurosci. Lett. 2019, 710, 132931.
- Witte, M.E.; Geurts, J.J.; de Vries, H.E.; van der Valk, P.; van Horssen, J. Mitochondrial dysfunction: A potential link between neuroinflammation and neurodegeneration? Mitochondrion 2010, 10, 411–418.
- Bajwa, E.; Pointer, C.B.; Klegeris, A. The Role of Mitochondrial Damage-Associated Molecular Patterns in Chronic Neuroinflammation. Mediat. Inflamm. 2019, 2019, 4050796.
- Trudler, D.; Nash, Y.; Frenkel, D. New insights on Parkinson’s disease genes: The link between mitochondria impairment and neuroinflammation. J. Neural. Transm. 2015, 122, 1409–1419.
- Riddell, N.; Crewther, S.G. Novel evidence for complement system activation in chick myopia and hyperopia models: A meta-analysis of transcriptome datasets. Science 2017, 7, 9719.
- Riddell, N.; Faou, P.; Murphy, M.; Giummarra, L.; Downs, R.A.; Rajapaksha, H.; Crewther, S.G. The retina/RPE proteome in chick myopia and hyperopia models: Commonalities with inherited and age-related ocular pathologies. Mol. Vis. 2017, 23, 872–888.
- Yin, J.; Reiman, E.M.; Beach, T.G.; Serrano, G.E.; Sabbagh, M.N.; Nielsen, M.; Caselli, R.J.; Shi, J. Effect of ApoE isoforms on mitochondria in Alzheimer disease. Neurology 2020, 94, e2404–e2411.
- Sun, F.; Deng, Y.; Han, X.; Liu, Q.; Zhang, P.; Manzoor, R.; Ma, H. A secret that underlies Parkinson’s disease: The damaging cycle. Neurochem. Int. 2019, 129, 104484.
- Franceschi, C.; Capri, M.; Monti, D.; Giunta, S.; Olivieri, F.; Sevini, F.; Panourgia, M.P.; Invidia, L.; Celani, L.; Scurti, M.; et al. Inflammaging and anti-inflammaging: A systemic perspective on aging and longevity emerged from studies in humans. Mech. Ageing Dev. 2007, 128, 92–105.
- Gyengesi, E.; Rangel, A.; Ullah, F.; Liang, H.; Niedermayer, G.; Asgarov, R.; Venigalla, M.; Gunawardena, D.; Karl, T.; Munch, G. Chronic Microglial Activation in the GFAP-IL6 Mouse Contributes to Age-Dependent Cerebellar Volume Loss and Impairment in Motor Function. Front. Neurosci. 2019, 13, 303.
- Tangestani, F.M.; Stough, C. A Review and Hypothesized Model of the Mechanisms That Underpin the Relationship between Inflammation and Cognition in the Elderly. Front. Aging Neurosci. 2019, 11, 56.
- Meszaros, A.; Molnar, K.; Nogradi, B.; Hernadi, Z.; Nyul-Toth, A.; Wilhelm, I.; Krizbai, I.A. Neurovascular Inflammaging in Health and Disease. Cells 2020, 9, 1614.
- Calabrese, V.; Santoro, A.; Monti, D.; Crupi, R.; Di Paola, R.; Latteri, S.; Cuzzocrea, S.; Zappia, M.; Giordano, J.; Calabrese, E.J. Aging and Parkinson’s Disease: Inflammaging, neuroinflammation and biological remodeling as key factors in pathogenesis. Free Radic. Biol. Med. 2018, 115, 80–91.
- Tronel, C.; Largeau, B.; Santiago Ribeiro, M.J.; Guilloteau, D.; Dupont, A.C.; Arlicot, N. Molecular Targets for PET Imaging of Activated Microglia: The Current Situation and Future Expectations. Int. J. Mol. Sci. 2017, 18, 802.
- Franceschi, C.; Bonafe, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; De Benedictis, G. Inflammaging. An evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci. 2000, 908, 244–254.
- Franceschi, C.; Campisi, J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J. Gerontol. Ser. A Biomed. Sci. Med. Sci. 2014, 69 (Suppl. S1), S4–S9.
- Beltran-Castillo, S.; Eugenin, J.; von Bernhardi, R. Impact of Aging in Microglia-Mediated D-Serine Balance in the CNS. Mediat. Inflamm. 2018, 2018, 7219732.
- Süβ, P.; Lana, A.J.; Schlachetzki, J.C.M. Chronic peripheral inflammation: A possible contributor to neurodegenerative diseases. Neural Regen. Res. 2021, 16, 1711–1714.
- Batatinha, H.A.P.; Diniz, T.A.; de Souza Teixeira, A.A.; Kruger, K.; Rosa-Neto, J.C. Regulation of autophagy as a therapy for immunosenescence-driven cancer and neurodegenerative diseases: The role of exercise. J. Cell. Physiol. 2019, 234, 14883–14895.
- Plaza-Zabala, A.; Sierra-Torre, V.; Sierra, A. Autophagy and Microglia: Novel Partners in Neurodegeneration and Aging. Int. J. Mol. Sci. 2017, 18, 598.
- Harry, G.J. Microglia during development and aging. Pharmacol. Ther. 2013, 139, 313–326.
- Tremblay, M.E.; Cookson, M.R.; Civiero, L. Glial phagocytic clearance in Parkinson’s disease. Mol. Neurodegener. 2019, 14, 16.
- Santoro, A.; Spinelli, C.C.; Martucciello, S.; Nori, S.L.; Capunzo, M.; Puca, A.A.; Ciaglia, E. Innate immunity and cellular senescence: The good and the bad in the developmental and aged brain. J. Leukoc. Biol. 2018, 103, 509–524.
- Fulop, T.; Larbi, A.; Dupuis, G.; Le Page, A.; Frost, E.H.; Cohen, A.A.; Witkowski, J.M.; Franceschi, C. Immunosenescence and inflammaging as two sides of the same coin: Friends or foes? Front. Immunol. 2018, 8, 1960.
- Dupré-Crochet, S.; Erard, M.; Nüβe, O. ROS production in phagocytes: Why, when, and where? J. Leukoc. Biol. 2013, 94, 657–670.
- Bournat, J.C.; Brown, C.W. Mitochondrial dysfunction in obesity. Curr. Opin. Endocrinol. Diabetes Obes. 2010, 17, 446–452.
- Krief, S.; Lonnqvist, F.; Raimbault, S.; Baude, B.; Van Spronsen, A.; Arner, P.; Strosberg, A.D.; Ricquier, D.; Emorine, L.J. Tissue distribution of beta 3-adrenergic receptor mRNA in man. J. Clin. Investig. 1993, 91, 344–349.
- Santiago, J.A.; Bottero, V.; Potashkin, J.A. Biological and Clinical Implications of Comorbidities in Parkinson’s Disease. Front. Aging Neurosci. 2017, 9, 394.
- Santiago, J.A.; Potashkin, J.A. System-based approaches to decode the molecular links in Parkinson’s disease and diabetes. Neurobiol. Dis. 2014, 72 Pt A, 84–91.
- Fuger, P.; Hefendehl, J.K.; Veeraraghavalu, K.; Wendeln, A.C.; Schlosser, C.; Obermuller, U.; Wegenast-Braun, B.M.; Neher, J.J.; Martus, P.; Kohsaka, S.; et al. Microglia turnover with aging and in an Alzheimer’s model via long-term in vivo single-cell imaging. Nat. Neurosci. 2017, 20, 1371–1376.
- Presta, I.; Vismara, M.; Novellino, F.; Donato, A.; Zaffino, P.; Scali, E.; Pirrone, K.C.; Spadea, M.F.; Malara, N.; Donato, G. Innate Immunity Cells and the Neurovascular Unit. Int. J. Mol. Sci. 2018, 19, 3856.
- Kapnick, S.M.; Pacheco, S.E.; McGuire, P.J. The emerging role of immune dysfunction in mitochondrial diseases as a paradigm for understanding immunometabolism. Metabolism 2018, 81, 97–112.
- Watson, E.; Davis, R.; Sue, C.M. New diagnostic pathways for mitochondrial disease. J. Transl. Genet. Genom. 2020, 4, 188–202.
- Wallace, D.C. Mitochondrial diseases in man and mouse. Science 1999, 283, 1482–1488.
- Wallace, D.C. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: A dawn for evolutionary medicine. Annu. Rev. Genet. 2005, 39, 359–407.
- Gorman, G.S.; Chinnery, P.F.; DiMauro, S.; Hirano, M.; Koga, Y.; McFarland, R.; Suomalainen, A.; Thorburn, D.R.; Zeviani, M.; Turnbull, D.M. Mitochondrial diseases. Nat. Rev. Dis. Prim. 2016, 2, 16080.
- Schapira, A.H. Mitochondrial disease. Lancet 2006, 368, 70–82.
- Schapira, A.H. Mitochondrial dysfunction in Parkinson’s disease. Cell Death Differ. 2007, 14, 1261–1266.
- Schapira, A.H.; Gegg, M. Mitochondrial contribution to Parkinson’s disease pathogenesis. Park. Dis. 2011, 2011, 159160.
- Edmonds, J.L. The otolaryngological manifestations of mitochondrial disease and the risk of neurodegeneration with infection. (Archives of Otolaryngology—Head & Neck Surgery). JAMA J. Am. Med. Assoc. 2002, 288, 296.
- Brand, M.D.; Nicholls, D.G. Assessing mitochondrial dysfunction in cells. Biochem. J. 2011, 435, 297–312.
- Deeb, W.; Nozile-Firth, K.; Okun, M.S. Parkinson’s disease: Diagnosis and appreciation of comorbidities. Handb. Clin. Neurol. 2019, 167, 257–277.
- Yu, J.W.; Walter, B.L. Addressing critical care gaps in inpatient Parkinson’s care–Minimizing the impact of comorbidities and developing new care delivery models. Park. Relat. Disord. 2022, 104, 121–122.
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. Lancet 2015, 386, 896–912.
- Tansey, M.G.; Wallings, R.L.; Houser, M.C.; Herrick, M.K.; Keating, C.E.; Joers, V. Inflammation and immune dysfunction in Parkinson disease. Nat. Rev. Immunol. 2022, 22, 657–673.
- Luna-Sánchez, M.; Bianchi, P.; Quintana, A. Mitochondria-Induced Immune Response as a Trigger for Neurodegeneration: A Pathogen from Within. Int. J. Mol. Sci. 2021, 22, 8523.
- Ferger, A.I.; Campanelli, L.; Reimer, V.; Muth, K.N.; Merdian, I.; Ludolph, A.C.; Witting, A. Effects of mitochondrial dysfunction on the immunological properties of microglia. J. Neuroinflamm. 2010, 7, 45.
- Fang, C.; Wei, X.; Wei, Y. Mitochondrial DNA in the regulation of innate immune responses. Protein Cell 2016, 7, 11–16.
- Takeshige, K.; Minakami, S. NADH- and NADPH-dependent formation of superoxide anions by bovine heart submitochondrial particles and NADH-ubiquinone reductase preparation. Biochem. J. 1979, 180, 129–135.
- Liu, X.L.; Wang, Y.D.; Yu, X.M.; Li, D.W.; Li, G.R. Mitochondria-mediated damage to dopaminergic neurons in Parkinson’s disease (Review). Int. J. Mol. Med. 2018, 41, 615–623.
- Ghosh, S.; Castillo, E.; Frias, E.S.; Swanson, R.A. Bioenergetic regulation of microglia. Glia 2018, 66, 1200–1212.
- Ahmad, M.H.; Fatima, M.; Mondal, A.C. Influence of microglia and astrocyte activation in the neuroinflammatory pathogenesis of Alzheimer’s disease: Rational insights for the therapeutic approaches. J. Clin. Neurosci. 2019, 59, 6–11.
- Protasoni, M.; Zeviani, M. Mitochondrial Structure and Bioenergetics in Normal and Disease Conditions. Int. J. Mol. Sci. 2021, 22, 586.
- Park, J.; Choi, H.; Min, J.S.; Park, S.J.; Kim, J.H.; Park, H.J.; Kim, B.; Chae, J.I.; Yim, M.; Lee, D.S. Mitochondrial dynamics modulate the expression of pro-inflammatory mediators in microglial cells. J. Neurochem. 2013, 127, 221–232.
- Reichenbach, J.; Schubert, R.; Horvath, R.; Petersen, J.; Futterer, N.; Malle, E.; Stumpf, A.; Gebhardt, B.R.; Koehl, U.; Schraven, B.; et al. Fatal neonatal-onset mitochondrial respiratory chain disease with T cell immunodeficiency. Pediatr. Res. 2006, 60, 321–326.
- Annesley, S.J.; Lay, S.T.; De Piazza, S.W.; Sanislav, O.; Hammersley, E.; Allan, C.Y.; Francione, L.M.; Bui, M.Q.; Chen, Z.P.; Ngoei, K.R.; et al. Immortalized Parkinson’s disease lymphocytes have enhanced mitochondrial respiratory activity. Dis. Model. Mech. 2016, 9, 1295–1305.
- Requejo-Aguilar, R.; Bolanos, J.P. Mitochondrial control of cell bioenergetics in Parkinson’s disease. Free Radic. Biol. Med. 2016, 100, 123–137.
- Scorziello, A.; Borzacchiello, D.; Sisalli, M.J.; Di Martino, R.; Morelli, M.; Feliciello, A. Mitochondrial Homeostasis and Signaling in Parkinson’s Disease. Front. Aging Neurosci. 2020, 12, 100.
- Garland, E.F.; Hartnell, I.J.; Boche, D. Microglia and Astrocyte Function and Communication: What Do We Know in Humans? Front. Neurosci. 2022, 16, 824888.
- Narendra, D.P.; Jin, S.; Tanaka, A.; Suen, D.; Gautier, C.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010, 8, e1000298.
- Ferger, A.I.; Campanelli, L.; Reimer, V.; Muth, K.N.; Merdian, I.; Ludolph, A.C.; Witting, A. Effects of mitochondrial dysfunction on the immunological properties of microglia. J. Neuroinflamm. 2010, 7, 45.
- Fang, C.; Wei, X.; Wei, Y. Mitochondrial DNA in the regulation of innate immune responses. Protein Cell 2016, 7, 11–16.
- Reichenbach, J.; Schubert, R.; Horvath, R.; Petersen, J.; Futterer, N.; Malle, E.; Stumpf, A.; Gebhardt, B.R.; Koehl, U.; Schraven, B.; et al. Fatal neonatal-onset mitochondrial respiratory chain disease with T cell immunodeficiency. Pediatr. Res. 2006, 60, 321–326.
- Annesley, S.J.; Fisher, P.R. Mitochondria in Health and Disease. Cells 2019, 8, 680.
- Annesley, S.J.; Lay, S.T.; De Piazza, S.W.; Sanislav, O.; Hammersley, E.; Allan, C.Y.; Francione, L.M.; Bui, M.Q.; Chen, Z.P.; Ngoei, K.R.; et al. Immortalized Parkinson’s disease lymphocytes have enhanced mitochondrial respiratory activity. Dis. Model. Mech. 2016, 9, 1295–1305.
- Requejo-Aguilar, R.; Bolanos, J.P. Mitochondrial control of cell bioenergetics in Parkinson’s disease. Free Radic. Biol. Med. 2016, 100, 123–137.
- Scorziello, A.; Borzacchiello, D.; Sisalli, M.J.; Di Martino, R.; Morelli, M.; Feliciello, A. Mitochondrial Homeostasis and Signaling in Parkinson’s Disease. Front. Aging Neurosci. 2020, 12, 100.
- Narendra, D.P.; Jin, S.; Tanaka, A.; Suen, D.; Gautier, C.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010, 8, e1000298.
- Pacelli, C.; Giguere, N.; Bourque, M.J.; Levesque, M.; Slack, R.S.; Trudeau, L.E. Elevated Mitochondrial Bioenergetics and Axonal Arborization Size Are Key Contributors to the Vulnerability of Dopamine Neurons. Curr. Biol. 2015, 25, 2349–2360.
- Luna-Sánchez, M.; Bianchi, P.; Quintana, A. Mitochondria-Induced Immune Response as a Trigger for Neurodegeneration: A Pathogen from Within. Int. J. Mol. Sci. 2021, 22, 8523.
- Elfawy, H.A.; Das, B. Crosstalk between mitochondrial dysfunction, oxidative stress, and age related neurodegenerative disease: Etiologies and therapeutic strategies. Life Sci. 2019, 218, 165–184.
- Protasoni, M.; Zeviani, M. Mitochondrial Structure and Bioenergetics in Normal and Disease Conditions. Int. J. Mol. Sci. 2021, 22, 586.
- Cheng, X.T.; Sheng, Z.H. Developmental regulation of microtubule-based trafficking and anchoring of axonal mitochondria in health and diseases. Dev. Neurobiol. 2021, 81, 284–299.
- Ray, B.; Bhat, A.; Mahalakshmi, A.M.; Tuladhar, S.; Bishir, M.; Mohan, S.K.; Veeraraghavan, V.P.; Chandra, R.; Essa, M.M.; Chidambaram, S.B.; et al. Mitochondrial and Organellar Crosstalk in Parkinson’s Disease. Am. Soc. Neurochem. 2021, 13, 17590914211028364.
- Trushina, E.; McMurray, C.T. Oxidative stress and mitochondrial dysfunction in neurodegenerative diseases. Neuroscience 2007, 145, 1233–1248.
- Mishra, A.K.; Dixit, A. Dopaminergic Axons: Key Recitalists in Parkinson’s Disease. Neurochem Res. 2022, 47, 234–248.
- Chen, C.; Turnbull, D.M.; Reeve, A.K. Mitochondrial Dysfunction in Parkinson’s Disease-Cause or Consequence? Biology 2019, 8, 38.
- Camandola, S.; Mattson, M.P. Brain metabolism in health, aging, and neurodegeneration. EMBO J. 2017, 36, 1474–1492.
- Landau, S.M.; Harvey, D.; Madison, C.M.; Reiman, E.M.; Foster, N.L.; Aisen, P.S.; Petersen, R.C.; Shaw, L.M.; Trojanowski, J.Q.; Jack, C.R., Jr.; et al. Comparing predictors of conversion and decline in mild cognitive impairment. Neurology 2010, 75, 230–238.