Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
The diagnosis and treatment of lung cancer (LC) is always a challenge. The difficulty in the decision of therapeutic schedule and diagnosis is directly related to intratumoral heterogeneity (ITH) in the progression of LC. It has been proven that most tumors emerge and evolve under the pressure of their living microenvironment, which involves genetic, immunological, metabolic, and therapeutic components.
- intratumoral heterogeneity
- lung cancer
1. Introduction
Intratumoral heterogeneity (ITH) is seen in most tumors during the investigation of the evolutionary trajectory of multiple cancer types. The factors affecting ITH in LC can be either genetic factors, pulmonary neoplastic microenvironmental factors, or metabolic factors. Both spatial and temporal ITH are influenced by the above three factors; therefore, both have emerged as typical characteristics in pulmonary neoplastic gene mutation, microenvironment, and metabolism.
While exploring the source of LC ITH, gene mutation was found to be the basis of spatiotemporal ITH in the pulmonary tumor microenvironment and LC metabolism. Recently, in a particular pathological type of lung cancer, remarkable genetic variation was found by multi-region genome-sequencing studies. The alterations were associated with not only anatomical position and stage of pulmonary neoplasms but also with distinct areas of the same tumor, which is often called spatial ITH. Additionally, the trait of the same nidus may vary prominently due to genetic factors and is referred to as temporal ITH. The dynamic headstream of variation of genetic factors is genetic alteration, such as gene mutation, which can occur in driver genes (can directly grant cancer cells superiority of survival and growth) and in passenger genes (cannot confer preponderance of selection). The form of gene mutation leading to ITH covers sequence insertion and deletion, single-nucleotide variants, and copy number variants [1].
From the perspective of therapy, the alteration of behavior correlated with the immunity of malignant pulmonary tumor should be seriously focused on. The characteristic immune response of pulmonary tumors can be recognized from the immunological ITH and tumor microenvironment (TME) ITH. Many elements in pulmonary TME vary both spatially and temporally, constructing a gigantic net of therapeutic schedules and pulmonary tumor ITH. In addition to the non-immune part, tumor immune microenvironment (TIME) ITH forms a landscape continuum based on spatiotemporal dimensionality, presented by affected immune cell function and immune factors. Multiple factors are secreted by pulmonary tumor cells and cells engaged in immune response progression.
Recognition of the relationship between metabolism and supervising gene changes in LC is a fundamental but inadequate process. Metabolic ITH is not only a factor affecting the variation of TME in LC but also a consequence of genetic ITH. Thus, the most appropriate dynamic detection of LC ITH can emerge from the perception of metabolic spatiotemporal ITH in LC. The acknowledgment of signal pathways associated with the alteration of metabolism and of the activation or suppression of typical key signal molecules is highly important. Two types of signal pathways exert a significant influence on metabolic ITH in LC, namely, phosphatidylinositol-3-kinase/protein kinase B/mammalian target of rapamycin (PI3K/AKT/mTOR) signal pathway and mitogen-activated protein kinases (MAPK) signal pathway.
In order to cure patients with LC and decrease their mortality, an integration concept should be formed by combining ITH with any therapeutic approach, no matter what mechanism the strategy is based on.
2. Genetic ITH in LC
2.1. Gene Mutation as a Source of Genetic ITH
Driver mutation in non-small cell lung cancer (NSCLC) occurs in EGFR, BRAF, and MET and is exclusively clonal and early, whereas subclonal driver mutation can occur in most instances, such as in PIK3CA, NF1, KRAS, TP53, and NOTCH family members. Two types of driver mutations assume distinct duties in tumor generation and therapies. When certainly targetable, an exclusively clonal driver mutation can be the direct result of targetable treatment across multiple positions of disease, subclonal driver mutation always appears subclonally or is absent in most regions. The latter can be clonal in a single region, where a large fraction of subclonal driver mutations accumulate [2][3] (Figure 1). In LC, a high subclonal mutation burden has been reported to be closely associated with human leukocyte antigen (HLA) loss of heterozygosity (LOH).
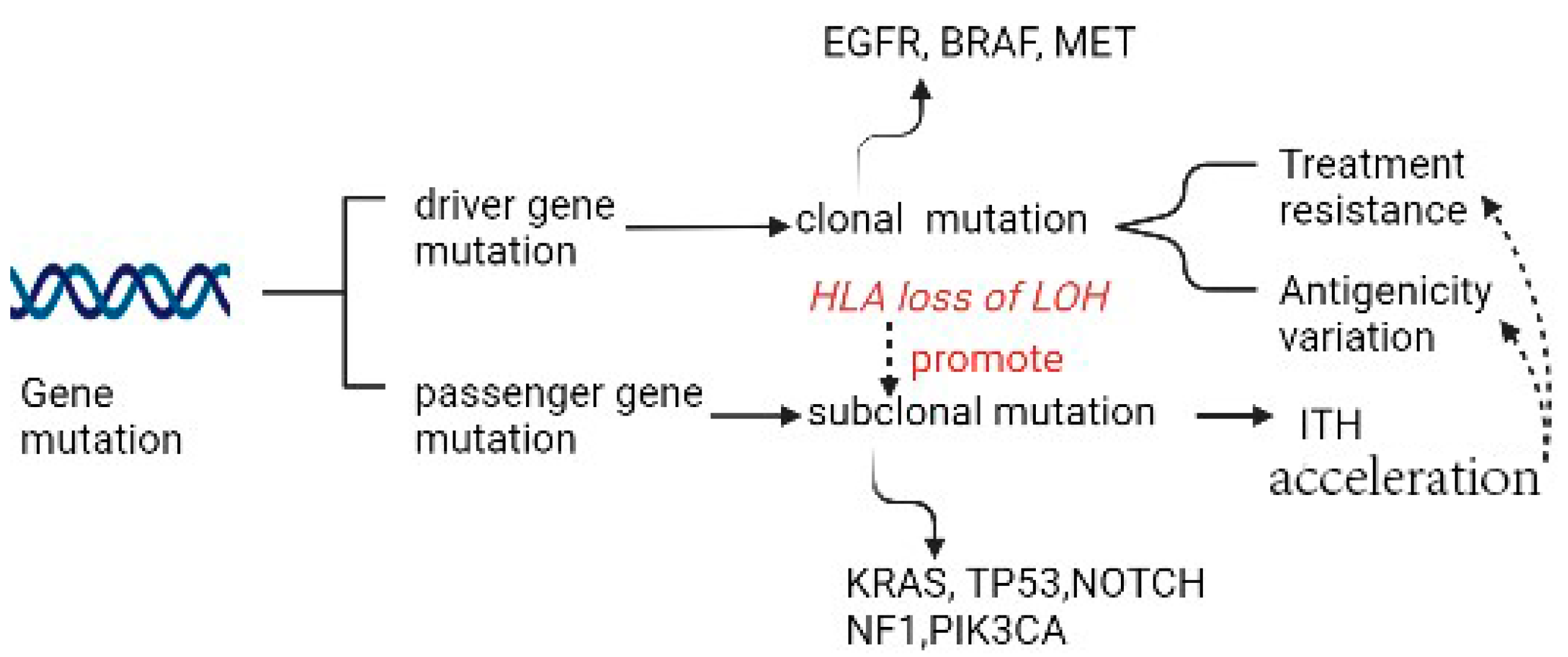
Figure 1. Gene mutation as the source of ITH.
In LC, the most frequent genes involved in pathogenesis in different pathological types have already been identified. For squamous cell carcinoma (SqCC) and adenocarcinoma (ADC), sequencing results have shown some differences; for example, the aberrations detected between ADC and SqCC are in KRAS (19% versus 2%), TP53 (44% versus 69%), and STK11 (21% versus 2%) [4]. For small cell lung cancer (SCLC), earlier findings had shown activating mutations of EGFR and KRAS and inactivating mutations of TP53 and Rb1 to be the most frequent [5]. Additionally, tumor-progressing factors related to genetic instability (mutations in DNA repair genes), treatment resistance, and antigenicity variation are of great importance [6].
Increasing evidence has shown TP53 and KRAS are necessary for ITH and to present various pathogenic variants in different populations. For example, the mutation frequencies of KRAS and TP53 in Iranian patients with LC are, respectively, lower and higher than in other populations [7]. In addition, Q808H in DDR2, F212L, and D186G in coding regions of TP53, three new putative pathogenic variants, have been first discovered in Iranian patients with LC [7]. The mutation frequencies of a consecutive population-based Swedish cohort have been proven to be similar to those observed in other Western populations, except for a notable high frequency (43%) of activating KRAS mutations among patients with lung adenocarcinoma [8]. In Western populations, a large fraction of ADCs harbors an activating mutation in KRAS [9]. Furthermore, the research showed patients who had the isolated KRAS mutation had a comparable overall survival rate to the wild-type group [10]. However, patients who had co-occurring mutations, specifically in TP53, had a poorer overall survival rate than both the wild-type group and the KRAS-only group [10]. Furthermore, a molecular subtype of KRAS-mutant LC with co-mutations in KEAP1/NFE2L2 was identified to result in a significantly shorter overall survival. Patients with concurrent mutations in KRAS and KEAP1/NFE2L2 had a shorter duration of therapy with platinum-based chemotherapy than other patients with KRAS-mutant lung cancer [10]. The mutation of either KRAS or TP53 had a tight connection with an enhanced PD-L1 expression [11][12]. A large and sustained clinical benefit was observed in KRASG12C/TP53mut, associated with a higher share of the highest PD-L1 expression levels (≥90%: 41.7% vs. 20.0% in KRASother) [13]. According to a large population-based case–control study conducted among Caucasians, Native Hawaiians residing on the island of Oahu, and Japanese, the substitution of arginine with proline at codon 72 of TP53 does not seem to perform a significant role in determining the risk of developing lung cancer [14]. The co-occurrence of TP53 and KRAS mutations was found to be associated with a higher mutation burden and was particularly enriched in the TH subset. This suggests that the dual mutation of TP53 and KRAS may have a synergistic and complementary effect on regulating immune biomarkers, leading to a responsive TME with adaptive immune resistance and increased immunogenicity [15].
Hence, the deepened exploration of LC gene mutation can help select appropriate therapy strategies. In preclinical studies with lung cancer cell lines expressing mutated TP53, the TP53 replacement strategy has improved chemotherapy and radiotherapy responses [16][17]. A meta-analysis of randomized controlled trials comparing combined therapy between immune checkpoint inhibitors (ICIs) and chemotherapy with chemotherapy alone showed that the addition of immune checkpoint inhibitors to chemotherapy may improve overall survival compared with chemotherapy alone [18]. Furthermore, the increasing evidence indicated that survival can benefit from adding immunotherapy (excluding checkpoint inhibitors) to conventional curative surgery or radiotherapy [19]. The application of ICIs, anti-PD1, and anti-PDL1 drugs has also been explored to select the best immunotherapy strategy for LC. A meta-analysis confirmed that the superiority of ICIs was over docetaxel in pretreated non-small-cell lung cancer patients and indicated a slight benefit from anti-PD-1 than from anti-PD-L1 inhibitors [20].
Gene mutation as the source of genetic ITH provided various signal targets for the development of therapeutic strategies and, meanwhile, made immunotherapy and other therapy strategies more flexible and specific to deal with treatment resistance due to mutation of various genes. Understanding the biology and natural history of LC is crucial for informing clinical management and therapeutic strategies. The discovery of significant ITH across various genomic types and similar tumor evolutionary trajectories for genetic changes has implications in this regard. Obtaining multiple biopsies and analyzing multiple genomic types may be necessary to capture the targetable events landscape accurately. Further studies are needed to identify the best combination of genomic types and regions to predict clinical outcomes, highlighting the importance of larger studies in the future.
2.2. Treatment Resistance Due to Specific Gene Mutation
When loss of phosphatase and tensin homolog (PTEN) occurs, resistance to PI3K inhibitors is seen. Activation of PIP3 phosphorylation accelerates the activity of protein kinase B, which is also called AKT. According to the activity of this central key enzyme, many pathways affecting cell survival, cell cycle progression, apoptosis, metabolism (such as protein synthesis, glycolysis, gluconeogenesis, and glucose uptake), cell proliferation, DNA repair, angiogenesis, and vesicle transport are likely to be suppressed or activated. Similarly, from the perspective of therapeutic resistance, owing to ITH in epidermal growth factor receptor (EGFR) or anaplastic lymphoma kinase (ALK), tyrosine kinase inhibitor (TKI) response rates are distinct across patients.
2.3. Antigenicity Variation Due to Specific Gene Mutation
Due to the loss of major histocompatibility complex (MHC) class I-coding genes in patients with lung cancer, pulmonary neoplasm can escape the scrutiny of the immune system [21]. Additionally, nonsynonymous mutations and indels in protein-coding genes create tumor neoantigens (TNAs) in addition to the normal antigens of pulmonary parenchyma cells and mesenchymal cells, which can be recognized and killed by tumor-specific CD8+ cytotoxic T lymphocytes (CTLs). According to previous clinical experience, TNAs due to gene mutation of KRAS in human gastrointestinal cancers have already been identified. It would be beneficial to harness the infiltration and cultivation of CTLs, in the course of treatment of tumors, which are triggered by KRAS mutation, which is considered one of the most malignant driver mutations. The prevalence of KRAS mutation can be high in most tumors, such as gastrointestinal cancers [22], as discussed previously for ADC.
2.4. Genomic Instability Due to Specific Gene Mutation
One type of gene mutation can influence both the possibility of genomic instability and the likelihood of TNA generation; it is called microsatellite instability (MSI), which benefits the extension or construction of short repetitive DNA sequences when DNA mismatch repair (MMR) genes appear to be defective [23]. MSI-high (MSI-H) pulmonary tumors are always associated with a high prevalence of generating TNAs, which can represent ITH in phenotype and be recognized by the specific immune system [24].
2.5. Relationship between Genetic ITH and Immunological ITH
Immunological ITH can be of three forms, namely, antigenicity, adjuvanticity, and immunoevasion [25]. Meanwhile, the neoplastic microenvironment can be changed by means of metabolic flexibility.
Immunological ITH in lung cancer is the manifestation of genetic ITH, according to the crucial degree of transcriptional ITH, of genes linked to immunity in spatial and temporal dimensions.
As a result, a huge stress exists on selection from the specific immune system, impacting the lung cancer cell clonal architecture, which has emerged as cancer clones or subclones. Lung cancer cells take advantage of the preceding mechanisms to escape the supervision and effect of the immune system. For instance, in lung cancer evolution, antigen expression can be suppressed in the form of limited antigenicity downstream, or antigen presentation can be impaired due to defects in molecular mechanisms [26][27]. Alterations can also be found in lung cancer cell adjuvanticity and in the establishment of a highly immunosuppressive TME [28][29].
3. ITH of Pulmonary Neoplastic Microenvironment
3.1. Immunological ITH and Pulmonary Neoplastic Microenvironment
Immunological ITH is one of the most principal factors affecting the neoplastic microenvironment, whereas the change in neoplastic microenvironment can accelerate or suppress the progress of genetic ITH, thereby influencing immunological ITH.
3.2. Other Neoplastic Microenvironmental Factors and TMH
Partial alterations or temporal variations in other TME factors, such as vascularization, stiffness, and inflammation, make a considerable contribution to neoplastic microenvironmental ITH [30].
ITH in TME (fused as tumor microenvironment heterogeneity (TMH)) provides a pulmonary tumor with the capability of drug resistance. Multiple growth factors and cytokines in TME induced by therapy can reprogram stromal cells toward protumor or antitumor phenotypes [31].
3.3. TIME ITH Affected by TME and Genetic ITH
The acknowledgment of the diversity of TIME (part of TME) due to TME factors and immunological ITH is of great significance so that new therapeutic targets can be found and their roles in patients receiving immune check-point blockade (ICB) therapy can be validated. The therapy is deemed to be a hindrance to the co-activities of the receptor and/or the ligand, such as cytotoxic T lymphocyte-associated antigen-4 (CTLA-4) and programmed cell death protein (PD-1). Recently, it has been found that vitamin E(VitE) can increase the antitumor efficacy of immune check-point therapy (ICT) (Figure 2). In this way, ITH occurs in TIME when protein tyrosine phosphatase SHP1 in dendritic cells (DCs) are inhibited. Then, DCs can enhance different levels of cross-presentation of tumor antigens, and different numbers of extracellular vesicles of DCs can trigger systemic antigen-specific T cell antitumor immunity in various degrees (Figure 2) [32].
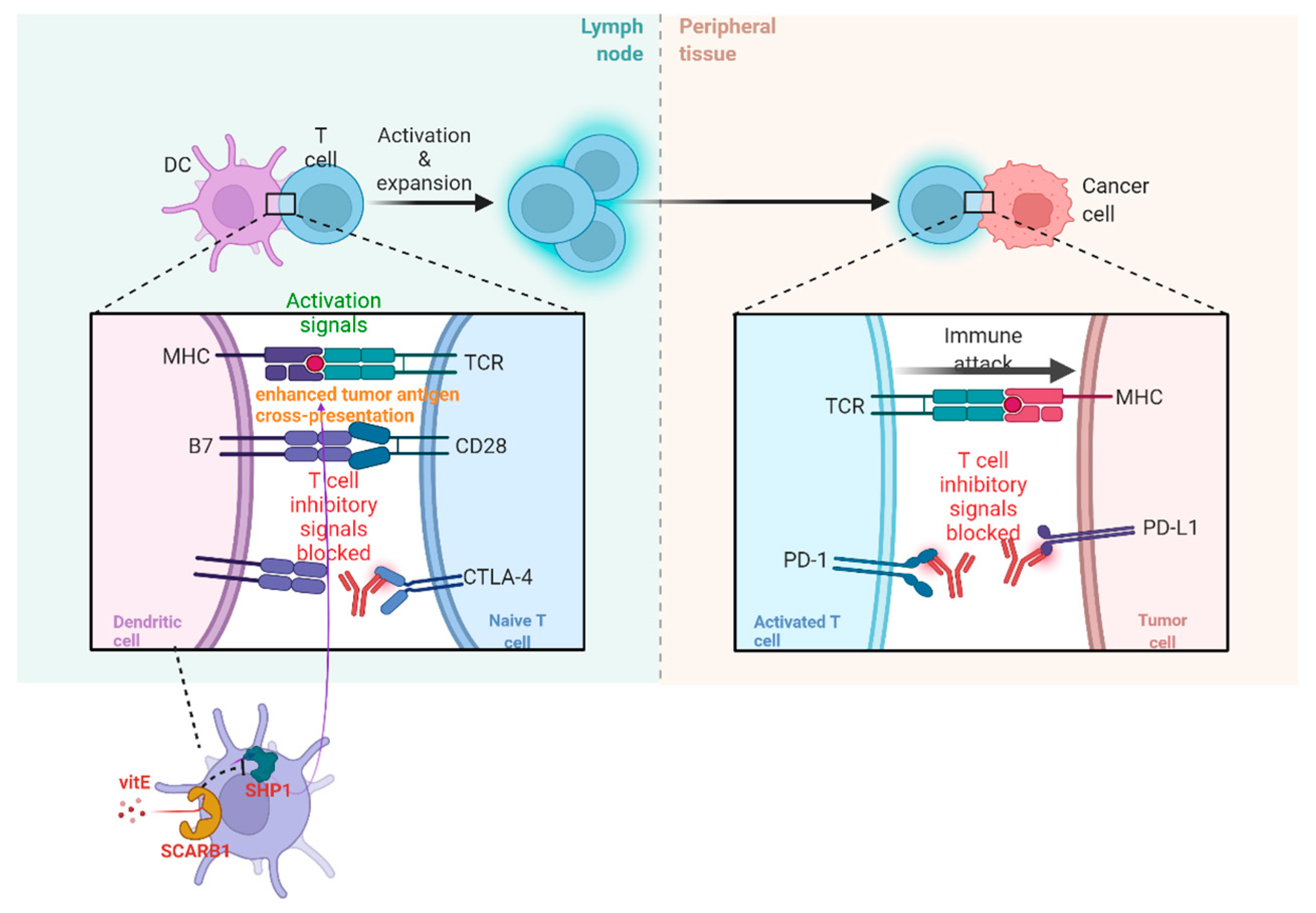
Figure 2. The operating principle of ICB and vitE.
In lung cancer, the source of TIME variation must be genetic ITH (Figure 3). The main pattern of manifestation of TIME ITH is in the alteration of cytokine production. As reported previously, cytokines whose expression is regulated by oncogenes are indispensable for the recruitment and phenotype of immune cells, especially myeloid immune cells. In advanced lung adenocarcinoma cases, KRAS G12D-triggered pancreatic ductal adenocarcinoma (PDAC) has been repeatedly demonstrated to secrete a large amount of granulocyte monocyte colony-stimulating factor (GM-CSF), suppressing immune function partially by improving the number of tumor-associated Gr-1+ CD11b+ myeloid cells. The immunosuppressive TIME established by the oncogene supports the development and proliferation of a variety of malignant tumors in addition to ADC, such as pancreatic neoplasia [33][34].

Figure 3. Relationship among genetic ITH, immunological ITH, and TME.
Furthermore, the secretion of tumor-derived chemokines, induced by specific oncogenes, is also important for the generation of TIME ITH (Figure 3). As is revealed by transcriptional analysis of tumor cells and in vitro DC migration assays in a mouse model of NSCLC with BRAF V600E mutation and PTEN-deficiency, reduction in chemokine CC motif ligand4 (Ccl4) production, which is deemed to be an efficient chemoattractant for multiple kinds of myeloid cells, including CD103+ DCs, could explain the undesirable infiltration of CD8+ T cells into TIME induced by the declined recruitment of CD103+ DCs.
Humoral factors in TME have also been confirmed to be momentous elements regulating TIME (Figure 3). For instance, pentraxin3 (PTX3) from mice, a significant regulator activating complement by means of reciprocity with factor H, can mediate monocyte recruitment and tumor-associated macrophage (TAM) phenotype to fulfill the purpose of repressing tumor growth [35]. Therefore, human PTX3 might affect the establishment of TIME similarly by the revelation of PTX3 promoter hypermethylation.
Tumor extrinsic factors can also perform a crucial role in triggering the profound change in the structure of TIME. For example, the non-immune stromal ingredient of TME can influence immune components. In NSCLC, BRAFV600E promotes the generation of interleukin (IL), such as IL-1α and IL-1β, hence repressing the recognition and killing capacity of tumor-specific CTLs, partially through the upregulation of PD-1 ligands, PD-L1, and PD-L2, and via secretion of cyclooxygenase (COX-2). A similar mechanism was also found in melanoma [36].
3.4. Immunoescape Affected by TIME
Immunoescape can also be facilitated by genetic ITH promoting the alterations in TIME. Genetic variations can occur due to mutations in genes regulating genomic immovability, such as MYC proto-oncogene (MYC), basic helix-loop-helix (bHLH) transcription factor, KRAS proto-oncogene GTPase (KRAS), and tumor protein p53 (TP53). Modification of these genes can elevate the abundance of immunosuppressive cell infiltration and/or organize the expression of coinhibitory molecules, such as PD-L1 or CTL exclusion. Genetic modifications can also make a difference in the differentiation of immune cells, such as of catenin beta 1 (CTNNB1), activation of which can exclude T cells [37].
3.5. Relationship of Immunoediting and ITH in LC
Cancer immunoediting is a dynamic process including three phases: elimination, equilibrium, and escape, whereby the immune system can not only protect against the development of tumors but also shape the cancer characters. A central principle of cancer immunoediting is associated with whether the T cell recognition of tumor antigens can promote the immunological elimination or sculpting of developing cancer. It has been proved that checkpoint blockade, such as the blocker targeting the PD-1/PD-L1 pathway, can effectively enhance T-cell-dependent immune-selective pressure. In addition to adaptive immunity and particularly T cells, innate immunity also performs a crucial role in tumor immunogenicity editing [38]. Hence, it can be concluded that immunoediting can produce variants with reduced immunogenicity, through which immunoediting facilitates immunological ITH.
Elimination is a phase of cancer immunoediting where the immune system can detect and destroy varieties of early tumors. The balance of tumor immunity is towards antitumor due to the upregulated expression of tumor antigens, FAS, TRAIL receptors, MHC class I on tumor cells and granzymes, IL-1, IL-12, TNF-α, IFN-α/β/γ, and perforin in TME. Therefore, the function of immunoediting also has a tight connection with TME. If TMH accelerates (such as an elevated spatial ITH of tumor-infiltrating lymphocytes (TILs) in LC), accumulated subclonal neoantigens mutations under the more immunoexhaustive and suppressive TME will contribute to more probability for tumor evasion [39]. The immunological and TME determine the destiny of both immunoediting and immune ITH. Tumors with low immune ITH can also escape, although the condition must be rigorous, including loss of HLA-LOH and immunoediting due to the high immunoselective pressure [40].
In the equilibrium phase of cancer immunoediting, the immune system sustains the tumor in a state of functional dormancy. The mechanism of this phase dynamically describes the development of tumor cells’ genetic and immunological ITH due to constant TME pressure. Tumor cell variants evolve towards the resistance of immune recognition (antigen defects and loss in antigen presentation) and enhancement of immunosuppression. In this phase, TME presents a balance between tumor-promoting cytokines (IL-10, IL-23) and antitumor cytokines (IL-12, IFN-γ). When NK cells and cytokines, such as IL-17A, IFN-α/β, and IL-4, are dispensable, the adaptive immune system will be required to involve in the maintenance of the functionally dormant state.
The mechanism of immune escape is fundamental for targeted immunotherapies. In this phase, tumor cells evade recognition of the immune system by loss of tumor antigens, co-stimulatory molecules or MHC class I, secrete cytokines TGF-β, IL-6, M-CSF, and VGEF, that increase angiogenesis and express molecules of immunosuppression (TDO, IDO, galectin-1/3/9, CD73, CD39, and PD-L1 adenosine receptors), survival (anti-apoptotic molecule bcl-2), and enhanced resistance (STAT-3). Furthermore, M2 macrophages, DCs, and myeloid-derived suppressor cells (MDSCs) can suppress the proliferation of CD8+ T cells or induce apoptosis by secreting IL-10 and TGF-β and express immunoregulatory molecules such as iNOS, IDO, and arginase. Regulatory T cells (Treg) can be generated by IDO-expressing DCs or MDSCs. T cells, including Treg cells, can express inhibitory receptors, such as LAG-3, Tim-3, PD-1, and CTLA-4, suppressing the antitumor immune response and promoting tumor outgrowth. In this phase, the balance is skewed toward tumor progression resulting from the immunosuppressive molecules and cytokines, such as PD-L1, IDO, VEGF, TGF-β, and IL-10. To dynamically recognize the ITH of TIME and appropriately combine blockers targeting different pathways closely associated with immunoevasion can revolutionize thinking about the therapeutic strategy of LC and approach to the essence of the pulmonary tumor.
This entry is adapted from the peer-reviewed paper 10.3390/cancers15102709
References
- Raynaud, F.; Mina, M.; Tavernari, D.; Ciriello, G. Pan-cancer inference of intra-tumor heterogeneity reveals associations with different forms of genomic instability. PLoS Genet. 2018, 14, e1007669.
- Young, K.; Minchom, A.; Larkin, J. BRIM-1, -2 and -3 trials: Improved survival with vemurafenib in metastatic melanoma patients with a BRAFV600E mutation. Futur. Oncol. 2012, 8, 499–507.
- Jamal-Hanjani, M.; Wilson, G.A.; McGranahan, N.; Birkbak, N.J.; Watkins, T.B.K.; Veeriah, S.; Shafi, S.; Johnson, D.H.; Mitter, R.; Rosenthal, R.; et al. Tracking the Evolution of Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 376, 2109–2121.
- Devarakonda, S.; Rotolo, F.; Tsao, M.; Lanc, I.; Brambilla, E.; Masood, A.; Olaussen, K.A.; Fulton, R.; Sakashita, S.; McLeer-Florin, A.; et al. Tumor Mutation Burden as a Biomarker in Resected Non–Small-Cell Lung Cancer. J. Clin. Oncol. 2018, 36, 2995–3006.
- Zito Marino, F.; Bianco, R.; Accardo, M.; Ronchi, A.; Cozzolino, I.; Morgillo, F.; Rossi, G.; Franco, R. Molecular Heterogeneity in Lung Cancer: From Mechanisms of Origin to Clinical Implications. Int. J. Med. Sci. 2019, 16, 981–989.
- Vitale, I.; Shema, E.; Loi, S.; Galluzzi, L. Intratumoral heterogeneity in cancer progression and response to immunotherapy. Nat. Med. 2021, 27, 212–224.
- Fathi, Z.; Mousavi, S.A.J.; Roudi, R.; Ghazi, F. Distribution of KRAS, DDR2, and TP53 gene mutations in lung cancer: An analysis of Iranian patients. PLoS ONE 2018, 13, e0200633.
- La Fleur, L.; Falk-Sörqvist, E.; Smeds, P.; Berglund, A.; Sundström, M.; Mattsson, J.S.; Brandén, E.; Koyi, H.; Isaksson, J.; Brunnström, H. Mutation patterns in a population-based non-small cell lung cancer cohort and prognostic impact of concomitant mutations in KRAS and TP53 or STK11. Lung Cancer 2019, 130, 50–58.
- Karachaliou, N.; Mayo, C.; Costa, C.; Magrí, I.; Gimenez-Capitan, A.; Molina-Vila, M.A.; Rosell, R. KRAS Mutations in Lung Cancer. Clin. Lung Cancer 2013, 14, 205–214.
- Arbour, K.C.; Jordan, E.; Kim, H.R.; Dienstag, J.; Yu, H.A.; Sanchez-Vega, F.; Lito, P.; Berger, M.; Solit, D.B.; Hellmann, M. Effects of Co-occurring Genomic Alterations on Outcomes in Patients with KRAS-Mutant Non–Small Cell Lung CancerCo-occurring Genomic Alterations in KRAS-Mutant NSCLC. Clin. Cancer Res. 2018, 24, 334–340.
- Van Gool, I.C.; Eggink, F.A.; Freeman-Mills, L.; Stelloo, E.; Marchi, E.; de Bruyn, M.; Palles, C.; Nout, R.A.; de Kroon, C.D.; Osse, E.M. POLE proofreading mutations elicit an antitumor immune response in endometrial cancer. Clin. Cancer Res. 2015, 21, 3347–3355.
- Caron, P.; Choudjaye, J.; Clouaire, T.; Bugler, B.; Daburon, V.; Aguirrebengoa, M.; Mangeat, T.; Iacovoni, J.S.; Álvarez-Quilón, A.; Cortés-Ledesma, F.; et al. Non-redundant Functions of ATM and DNA-PKcs in Response to DNA Double-Strand Breaks. Cell Rep. 2015, 13, 1598–1609.
- Garon, E.B.; Rizvi, N.A.; Hui, R.; Leighl, N.; Balmanoukian, A.S.; Eder, J.P.; Patnaik, A.; Aggarwal, C.; Gubens, M.; Horn, L.; et al. Pembrolizumab for the Treatment of Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 372, 2018–2028.
- Pierce, L.M.; Sivaraman, L.; Chang, W.; Lum, A.; Donlon, T.; Seifried, A.; Wilkens, L.R.; Lau, A.F.; Le Marchand, L. Relationships of TP53 codon 72 and HRAS1 polymorphisms with lung cancer risk in an ethnically diverse population. Cancer Epidemiol. Biomark. Prev. 2000, 9, 1199–1204.
- Dong, Z.-Y.; Zhong, W.-Z.; Zhang, X.-C.; Su, J.; Xie, Z.; Liu, S.-Y.; Tu, H.-Y.; Chen, H.-J.; Sun, Y.-L.; Zhou, Q. Potential predictive value of TP53 and KRAS mutation status for response to PD-1 blockade immunotherapy in lung adenocarcinoma. Clin. Cancer Res. 2017, 23, 3012–3024.
- Fujiwara, T.; Cai, D.W.; Georges, R.N.; Mukhopadhyay, T.; Grimm, E.A.; Roth, J.A. Therapeutic Effect of a Retroviral Wild-Type p53 Expression Vector in an Orthotopic Lung Cancer Model. Gynecol. Oncol. J. Natl. Cancer Inst. 1994, 86, 1458–1462.
- Roth, J.; Nguyen, D.; Lawrence, D.; Kemp, B.; Carrasco, C.; Ferson, D.; Hong, W.; Komaki, R.; Lee, J.; Nesbitt, J. Retrovirus–mediated wild–type P53 gene transfer to tumors of patients with lung cancer. Nat. Med. 1996, 2, 985–991.
- Petrelli, F.; Ferrara, R.; Signorelli, D.; Ghidini, A.; Proto, C.; Roudi, R.; Sabet, M.N.; Facelli, S.; Garassino, M.C.; Luciani, A.; et al. Immune checkpoint inhibitors and chemotherapy in first-line NSCLC: A meta-analysis. Immunotherapy 2021, 13, 621–631.
- Zhu, J.; Yuan, Y.; Wan, X.; Yin, D.; Li, R.; Chen, W.; Suo, C.; Song, H. Immunotherapy (excluding checkpoint inhibitors) for stage I to III non-small cell lung cancer treated with surgery or radiotherapy with curative intent. Cochrane Database Syst. Rev. 2017, 12, CD011300.
- Tartarone, A.; Roviello, G.; Lerose, R.; Roudi, R.; Aieta, M.; Zoppoli, P.; Reck, M.; Borghaei, H.; O’byrne, K.J.; Santoni, M.; et al. Anti-PD-1 versus anti-PD-L1 therapy in patients with pretreated advanced non-small-cell lung cancer: A meta-analysis. Futur. Oncol. 2019, 15, 2423–2433.
- Montesion, M.; Murugesan, K.; Jin, D.X.; Sharaf, R.; Sanchez, N.; Guria, A.; Minker, M.; Li, G.; Fisher, V.; Sokol, E.S.; et al. Somatic HLA Class I Loss Is a Widespread Mechanism of Immune Evasion Which Refines the Use of Tumor Mutational Burden as a Biomarker of Checkpoint Inhibitor Response. Cancer Discov. 2021, 11, 282–292.
- Tran, E.; Ahmadzadeh, M.; Lu, Y.-C.; Gros, A.; Turcotte, S.; Robbins, P.F.; Gartner, J.J.; Zheng, Z.; Li, Y.F.; Ray, S.; et al. Immunogenicity of somatic mutations in human gastrointestinal cancers. Science 2015, 350, 1387–1390.
- Hause, R.J.; Pritchard, C.C.; Shendure, J.; Salipante, S.J. Classification and characterization of microsatellite instability across 18 cancer types. Nat. Med. 2016, 22, 1342–1350.
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413.
- Losic, B.; Craig, A.J.; Villacorta-Martin, C.; Martins-Filho, S.N.; Akers, N.; Chen, X.; Ahsen, M.E.; von Felden, J.; Labgaa, I.; Dʹavola, D.; et al. Intratumoral heterogeneity and clonal evolution in liver cancer. Nat. Commun. 2020, 11, 291.
- Rosenthal, R.; Cadieux, E.L.; Salgado, R.; Bakir, M.A.; Moore, D.A.; Hiley, C.T.; Lund, T.; Tanić, M.; Reading, J.L.; Joshi, K.; et al. Neoantigen-directed immune escape in lung cancer evolution. Nature 2019, 567, 479–485.
- McGranahan, N.; Rosenthal, R.; Hiley, C.T.; Rowan, A.J.; Watkins, T.B.K.; Wilson, G.A.; Birkbak, N.J.; Veeriah, S.; Van Loo, P.; Herrero, J.; et al. Allele-Specific HLA Loss and Immune Escape in Lung Cancer Evolution. Cell 2017, 171, 1259–1271.e11.
- Senovilla, L.; Vitale, I.; Martins, I.; Tailler, M.; Pailleret, C.; Michaud, M.; Galluzzi, L.; Adjemian, S.; Kepp, O.; Niso-Santano, M.; et al. An Immunosurveillance Mechanism Controls Cancer Cell Ploidy. Science 2012, 337, 1678–1684.
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550.
- Dagogo-Jack, I.; Shaw, A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94.
- Labrie, M.; Brugge, J.S.; Mills, G.B.; Zervantonakis, I.K. Therapy resistance: Opportunities created by adaptive responses to targeted therapies in cancer. Nat. Rev. Cancer 2022, 22, 323–339.
- Yuan, X.; Duan, Y.; Xiao, Y.; Sun, K.; Qi, Y.; Zhang, Y.; Ahmed, Z.; Moiani, D.; Yao, J.; Li, H.; et al. Vitamin E Enhances Cancer Immunotherapy by Reinvigorating Dendritic Cells via Targeting Checkpoint SHP. Cancer Discov. 2022, 12, 1742–1759.
- Pylayeva-Gupta, Y.; Lee, K.E.; Hajdu, C.H.; Miller, G.; Bar-Sagi, D. Oncogenic Kras-Induced GM-CSF Production Promotes the Development of Pancreatic Neoplasia. Cancer Cell 2012, 21, 836–847.
- Bayne, L.J.; Beatty, G.L.; Jhala, N.; Clark, C.E.; Rhim, A.D.; Stanger, B.Z.; Vonderheide, R.H. Tumor-Derived Granulocyte-Macrophage Colony-Stimulating Factor Regulates Myeloid Inflammation and T Cell Immunity in Pancreatic Cancer. Cancer Cell 2012, 21, 822–835.
- Bonavita, E.; Gentile, S.; Rubino, M.; Maina, V.; Papait, R.; Kunderfranco, P.; Greco, C.; Feruglio, F.; Molgora, M.; Laface, I.; et al. PTX3 Is an Extrinsic Oncosuppressor Regulating Complement-Dependent Inflammation in Cancer. Cell 2015, 160, 700–714.
- Khalili, J.S.; Liu, S.; Rodriguez-Cruz, T.G.; Whittington, M.; Wardell, S.; Liu, C.; Zhang, M.; Cooper, Z.A.; Frederick, D.T.; Li, Y.; et al. Oncogenic BRAF(V600E) promotes stromal cell-mediated immunosuppression via induction of interleukin-1 in melanoma. Clin. Cancer Res. 2012, 18, 5329–5340.
- Spranger, S.; Gajewski, T.F. Impact of oncogenic pathways on evasion of antitumour immune responses. Nat. Rev. Cancer 2018, 18, 139–147.
- O’Sullivan, T.; Saddawi-Konefka, R.; Vermi, W.; Koebel, C.M.; Arthur, C.; White, J.M.; Uppaluri, R.; Andrews, D.; Ngiow, S.F.; Teng, M.; et al. Cancer immunoediting by the innate immune system in the absence of adaptive immunity. J. Exp. Med. 2012, 209, 1869–1882.
- Joshi, K.; TRACERx Consortium; De Massy, M.R.; Ismail, M.; Reading, J.; Uddin, I.; Woolston, A.; Hatipoglu, E.; Oakes, T.; Rosenthal, R.; et al. Spatial heterogeneity of the T cell receptor repertoire reflects the mutational landscape in lung cancer. Nat. Med. 2019, 25, 1549–1559.
- Nguyen, P.H.D.; Ma, S.; Phua, C.Z.J.; Kaya, N.A.; Lai, H.L.H.; Lim, C.J.; Lim, J.Q.; Wasser, M.; Lai, L.; Tam, W.L.; et al. Intratumoural immune heterogeneity as a hallmark of tumour evolution and progression in hepatocellular carcinoma. Nat. Commun. 2021, 12, 227.
This entry is offline, you can click here to edit this entry!