The endocannabinoid system (ECS) is an important signaling pathway that involves the binding of lipid ligands, known as cannabinoids, to cannabinoid receptors, and it encompasses, as well, the metabolic enzymes of endocannabinoids. Endocannabinoid signaling plays crucial roles in human physiology in the function of multiple systems. The two cannabinoid receptors, CB1 and CB2, are cell membrane proteins that interact with both exogenous and endogenous bioactive lipid ligands, or endocannabinoids. Recent evidence has established that endocannabinoid signaling operates within the human kidney, as well as suggests the important role it plays in multiple renal pathologies.
- kidney
- nephron
- cannabinoid receptor 1
- podocyte
- chronic kidney disease
- fibrosis
- acute kidney injury
1. The Endocannabinoid System and Chronic Kidney Disease
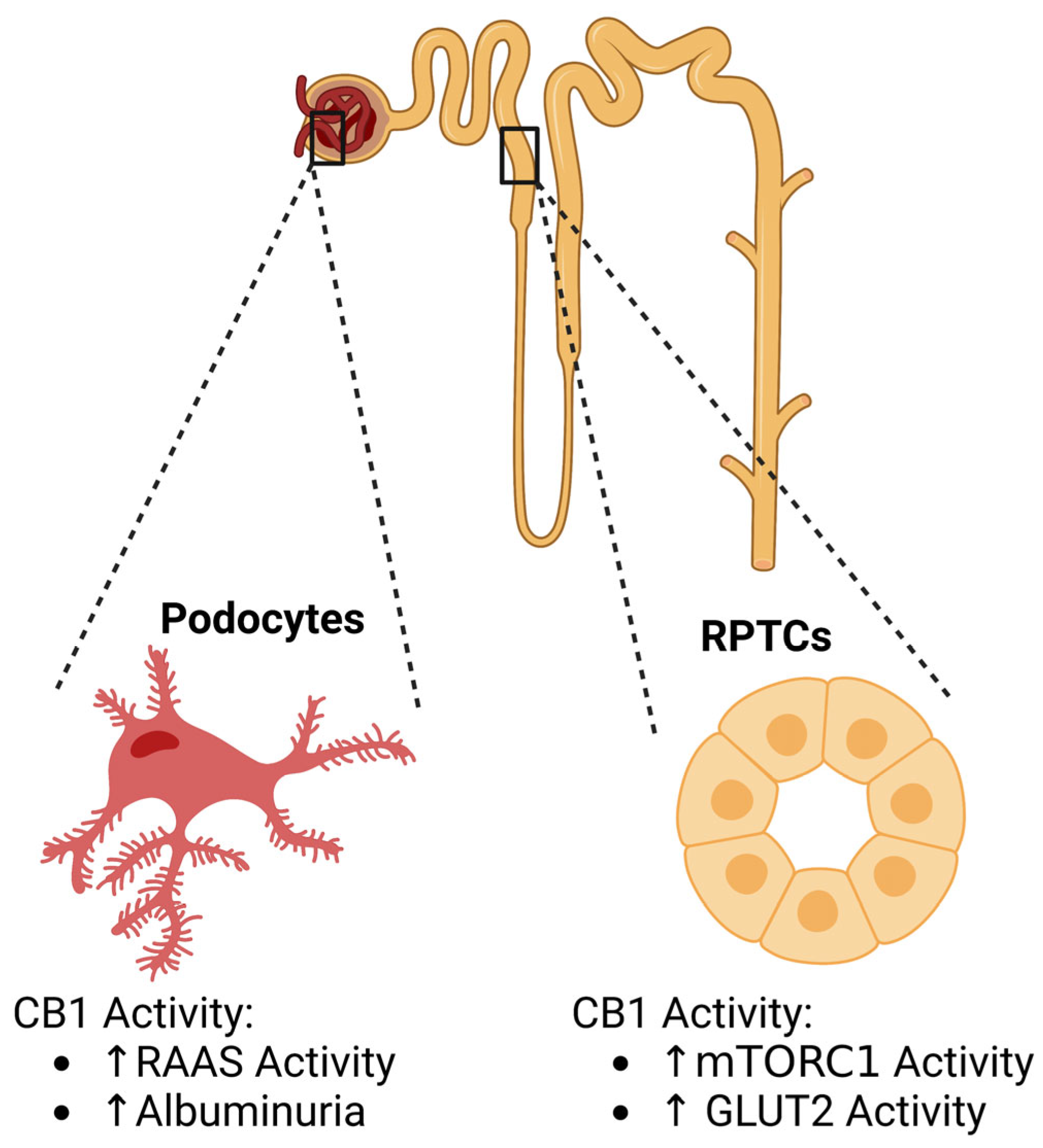
1.1. Diabetic Kidney Disease
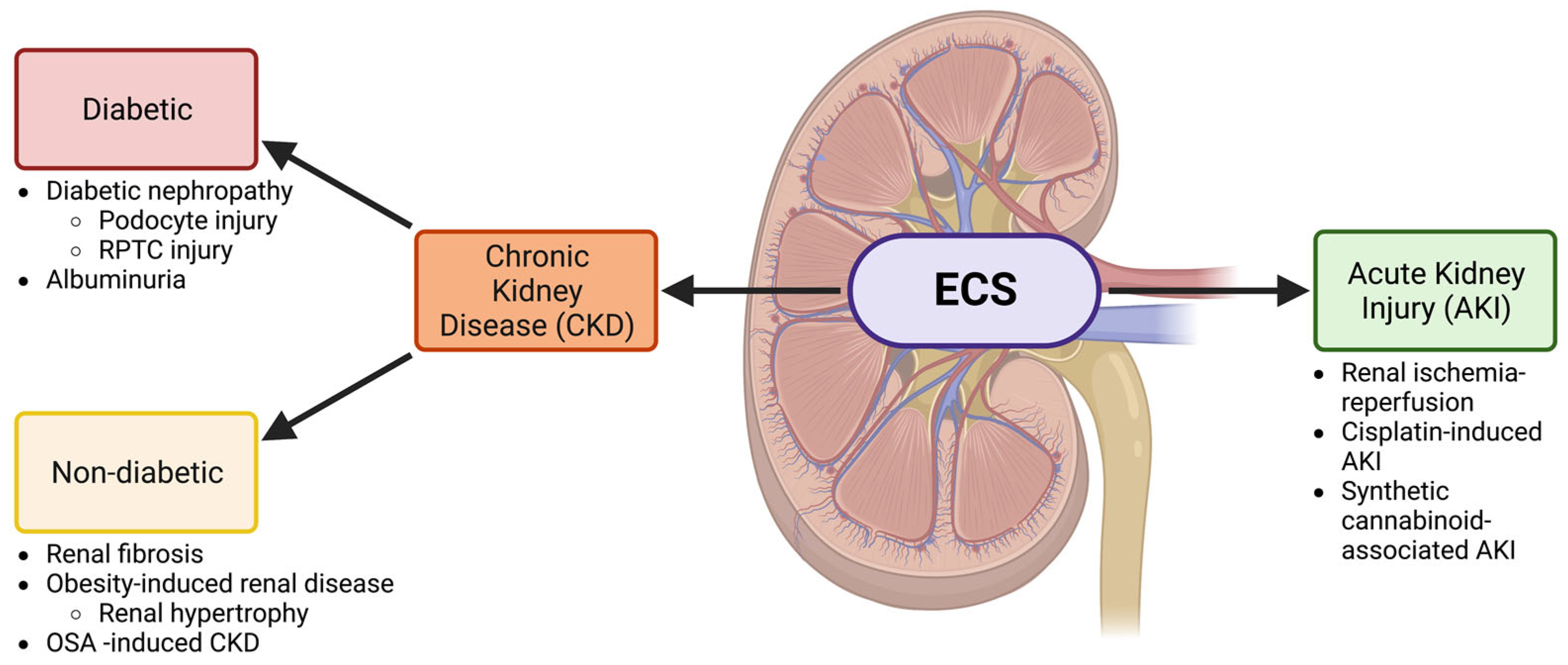
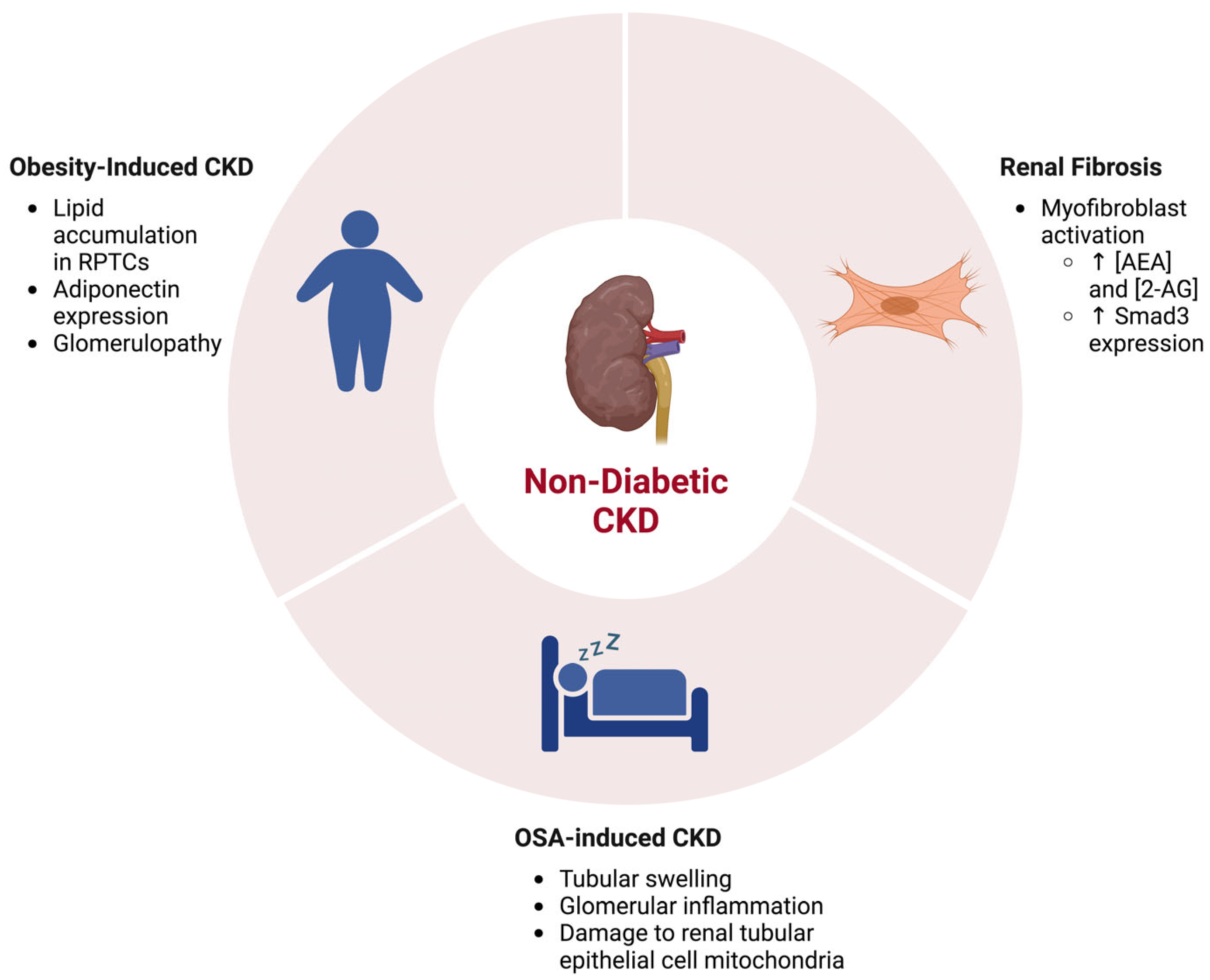
1.2. The Endocannabinoid System in Renal Fibrosis and Other Non-Diabetic Chronic Kidney Diseases
2. The Endocannabinoid System and Acute Kidney Injury
This entry is adapted from the peer-reviewed paper 10.3390/cells12101419
References
- Chua, J.T.; Argueta, D.A.; DiPatrizio, N.V.; Kovesdy, C.P.; Vaziri, N.D.; Kalantar-Zadeh, K.; Moradi, H. Endocannabinoid system and the kidneys: From renal physiology to injury and disease. Cannabis Cannabinoid Res. 2019, 4, 10–20.
- Jourdan, T.; Szanda, G.; Rosenberg, A.Z.; Tam, J.; Earley, B.J.; Godlewski, G.; Cinar, R.; Liu, Z.; Liu, J.; Ju, C.; et al. Overactive cannabinoid 1 receptor in podocytes drives type 2 diabetic nephropathy. Proc. Natl. Acad. Sci. USA 2014, 111, E5420–E5428.
- de Boer, I.H.; Khunti, K.; Sadusky, T.; Tuttle, K.R.; Neumiller, J.J.; Rhee, C.M.; Rosas, S.E.; Rossing, P.; Bakris, G. Diabetes management in chronic kidney disease: A consensus report by the american diabetes association (ADA) and kidney disease: Improving global outcomes (KDIGO). Diabetes Care 2022, 45, 3075–3090.
- Drori, A.; Permyakova, A.; Hadar, R.; Udi, S.; Nemirovski, A.; Tam, J. Cannabinoid-1 receptor regulates mitochondrial dynamics and function in renal proximal tubular cells. Diabetes Obes. Metab. 2019, 21, 146–159.
- Larrinaga, G.; Varona, A.; Pérez, I.; Sanz, B.; Ugalde, A.; Cándenas, M.L.; Pinto, F.M.; Gil, J.; López, J.I. Expression of cannabinoid receptors in human kidney. Histol. Histopathol. 2010, 25, 1133–1138.
- Bridgewater, D.J.; Ho, J.; Sauro, V.; Matsell, D.G. Insulin-like growth factors inhibit podocyte apoptosis through the PI3 kinase pathway. Kidney Int. 2005, 67, 1308–1314.
- Rohbeck, E.; Eckel, J.; Romacho, T. Cannabinoid receptors in metabolic regulation and diabetes. Physiology 2021, 36, 102–113.
- Barutta, F.; Corbelli, A.; Mastrocola, R.; Gambino, R.; Di Marzo, V.; Pinach, S.; Rastaldi, M.P.; Perin, P.C.; Gruden, G. Cannabinoid receptor 1 blockade ameliorates albuminuria in experimental diabetic nephropathy. Diabetes 2010, 59, 1046–1054.
- Nam, D.H.; Lee, M.H.; Kim, J.E.; Song, H.K.; Kang, Y.S.; Lee, J.E.; Kim, H.W.; Cha, J.J.; Hyun, Y.Y.; Kim, S.H.; et al. Blockade of cannabinoid receptor 1 improves insulin resistance, lipid metabolism, and diabetic nephropathy in db/db mice. Endocrinology 2012, 153, 1387–1396.
- Barutta, F.; Bellini, S.; Mastrocola, R.; Gambino, R.; Piscitelli, F.; di Marzo, V.; Corbetta, B.; Vemuri, V.K.; Makriyannis, A.; Annaratone, L.; et al. Reversal of albuminuria by combined AM6545 and perindopril therapy in experimental diabetic nephropathy. Br. J. Pharmacol. 2018, 175, 4371–4385.
- Jourdan, T.; Park, J.K.; Varga, Z.V.; Pálóczi, J.; Coffey, N.J.; Rosenberg, A.Z.; Godlewski, G.; Cinar, R.; Mackie, K.; Pacher, P.; et al. Cannabinoid-1 receptor deletion in podocytes mitigates both glomerular and tubular dysfunction in a mouse model of diabetic nephropathy. Diabetes Obes. Metab. 2018, 20, 698–708.
- Raja, P.; Maxwell, A.P.; Brazil, D.P. The potential of albuminuria as a biomarker of diabetic complications. Cardiovasc. Drugs Ther. 2021, 35, 455–466.
- Kogot-Levin, A.; Hinden, L.; Riahi, Y.; Israeli, T.; Tirosh, B.; Cerasi, E.; Mizrachi, E.B.; Tam, J.; Mosenzon, O.; Leibowitz, G. Proximal tubule mTORC1 is a central player in the pathophysiology of diabetic nephropathy and its correction by SGLT2 inhibitors. Cell Rep. 2020, 32, 107954.
- Hinden, L.; Ahmad, M.; Hamad, S.; Nemirovski, A.; Szanda, G.; Glasmacher, S.; Kogot-Levin, A.; Abramovitch, R.; Thorens, B.; Gertsch, J.; et al. Opposite physiological and pathological mTORC1-mediated roles of the CB1 receptor in regulating renal tubular function. Nat. Commun. 2022, 13, 1783–1788.
- Sampaio, L.S.; Taveira Da Silva, R.; Lima, D.; Sampaio, C.L.C.; Iannotti, F.A.; Mazzarella, E.; Di Marzo, V.; Vieyra, A.; Reis, R.A.M.; Einicker-Lamas, M. The endocannabinoid system in renal cells: Regulation of Na(+) transport by CB1 receptors through distinct cell signalling pathways. Br. J. Pharmacol. 2015, 172, 4615–4625.
- Jenkin, K.A.; McAinch, A.J.; Grinfeld, E.; Hryciw, D.H. Role for cannabinoid receptors in human proximal tubular hypertrophy. Cell. Physiol. Biochem. 2010, 26, 879–886.
- Udi, S.; Hinden, L.; Earley, B.; Drori, A.; Reuveni, N.; Hadar, R.; Cinar, R.; Nemirovski, A.; Tam, J. Proximal tubular cannabinoid-1 receptor regulates obesity-induced CKD. J. Am. Soc. Nephrol. 2017, 28, 3518–3532.
- Anders, H.; Huber, T.B.; Isermann, B.; Schiffer, M. CKD in diabetes: Diabetic kidney disease versus nondiabetic kidney disease. Nat. Rev. Nephrol. 2018, 14, 361–377.
- Prakoura, N.; Hadchouel, J.; Chatziantoniou, C. Novel targets for therapy of renal fibrosis. J. Histochem. Cytochem. 2019, 67, 701–715.
- Lecru, L.; Desterke, C.; Grassin-Delyle, S.; Chatziantoniou, C.; Vandermeersch, S.; Devocelle, A.; Vernochet, A.; Ivanovski, N.; Ledent, C.; Ferlicot, S.; et al. Cannabinoid receptor 1 is a major mediator of renal fibrosis. Kidney Int. 2015, 88, 72–84.
- Golosova, D.; Levchenko, V.; Kravtsova, O.; Palygin, O.; Staruschenko, A. Acute and long-term effects of cannabinoids on hypertension and kidney injury. Sci. Rep. 2022, 12, 6080–6086.
- Sato, M.; Muragaki, Y.; Saika, S.; Roberts, A.B.; Ooshima, A. Targeted disruption of TGF-beta1/Smad3 signaling protects against renal tubulointerstitial fibrosis induced by unilateral ureteral obstruction. J. Clin. Investig. 2003, 112, 1486–1494.
- Wu, W.; Wang, X.; Yu, X.; Lan, H. Smad3 signatures in renal inflammation and fibrosis. Int. J. Biol. Sci. 2022, 18, 2795–2806.
- Meng, X.; Tang, P.M.; Li, J.; Lan, H.Y. TGF-β/smad signaling in renal fibrosis. Front. Physiol. 2015, 6, 82.
- Chen, D.; Tang, H.; Jiang, H.; Sun, L.; Zhao, W.; Qian, F. ACPA alleviates bleomycin-induced pulmonary fibrosis by inhibiting TGF-β-Smad2/3 signaling-mediated lung fibroblast activation. Front. Pharmacol. 2022, 13, 835979.
- Yoshinaga, T.; Uwabe, K.; Naito, S.; Higashino, K.; Nakano, T.; Numata, Y.; Kihara, A. AM251 suppresses epithelial-mesenchymal transition of renal tubular epithelial cells. PLoS ONE 2016, 11, e0167848.
- Miranda, K.; Mehrpouya-Bahrami, P.; Nagarkatti, P.S.; Nagarkatti, M. Cannabinoid receptor 1 blockade attenuates obesity and adipose tissue type 1 inflammation through miR-30e-5p regulation of delta-like-4 in macrophages and consequently downregulation of Th1 cells. Front. Immunol. 2019, 10, 1049.
- Janiak, P.; Poirier, B.; Bidouard, J.; Cadrouvele, C.; Pierre, F.; Gouraud, L.; Barbosa, I.; Dedio, J.; Maffrand, J.P.; Le Fur, G.; et al. Blockade of cannabinoid CB1 receptors improves renal function, metabolic profile, and increased survival of obese Zucker rats. Kidney Int. 2007, 72, 1345–1357.
- Wang, Y.; Yu, Y.; Zhang, H.; Chen, C.; Wan, H.; Chen, Y.; Xia, F.; Yu, S.; Wang, N.; Ye, L.; et al. Cardiovascular and renal burdens among patients with MAFLD and NAFLD in China. Front. Endocrinol. 2022, 13, 968766.
- Udi, S.; Hinden, L.; Ahmad, M.; Drori, A.; Iyer, M.R.; Cinar, R.; Herman-Edelstein, M.; Tam, J. Dual inhibition of cannabinoid CB1 receptor and inducible NOS attenuates obesity-induced chronic kidney disease. Br. J. Pharmacol. 2020, 177, 110–127.
- Abuyassin, B.; Badran, M.; Ayas, N.T.; Laher, I. Intermittent hypoxia causes histological kidney damage and increases growth factor expression in a mouse model of obstructive sleep apnea. PLoS ONE 2018, 13, e0192084.
- Dou, Z.; Gao, X.; Jia, Y.; Chen, J.; Yang, J.; Chen, Y.; Wu, S.J.; Liu, T.; Wang, M.T.; Yang, C.; et al. CB1 receptor antagonist rimonabant protects against chronic intermittent hypoxia-induced bone metabolism disorder and destruction in rats. Sleep Breath. 2020, 24, 1441–1449.
- Lee, Y.; Hung, S.; Wang, H.; Lin, C.; Wang, H.; Chen, S.; Chang, M.Y.; Ho, L.C.; Chen, Y.T.; Liou, H.H.; et al. Sleep apnea and the risk of chronic kidney disease: A nationwide population-based cohort study. Sleep 2015, 38, 213–221.
- Zhao, L.; Liu, T.; Dou, Z.; Wang, M.; Hu, Z.; Wang, B. CB1 receptor antagonist rimonabant protects against chronic intermittent hypoxia-induced renal injury in rats. BMC Nephrol. 2021, 22, 153.
- Malek, M.; Nematbakhsh, M. Renal ischemia/reperfusion injury; from pathophysiology to treatment. J. Renal Inj. Prev. 2015, 4, 20–27.
- Zhang, Z.; Zhao, J.; Dong, W.; Remer, E.; Li, J.; Demirjian, S.; Zabell, J.; Campbell, S.C. Acute kidney injury after partial nephrectomy: Role of parenchymal mass reduction and ischemia and impact on subsequent functional recovery. Eur. Urol. 2016, 69, 745–752.
- Pacher, P.; Haskó, G. Endocannabinoids and cannabinoid receptors in ischaemia-reperfusion injury and preconditioning. Br. J. Pharmacol. 2008, 153, 252–262.
- Sampaio, L.S.; Iannotti, F.A.; Veneziani, L.; Borelli-Tôrres, R.T.; De Maio, F.; Piscitelli, F.; Reis, R.A.M.; Di Marzo, V.; Einicker-Lamas, M. Experimental ischemia/reperfusion model impairs endocannabinoid signaling and Na+/K+ ATPase expression and activity in kidney proximal tubule cells. Biochem. Pharmacol. 2018, 154, 482–491.
- Moradi, H.; Oveisi, F.; Khanifar, E.; Moreno-Sanz, G.; Vaziri, N.D.; Piomelli, D. Increased renal 2-arachidonoylglycerol level is associated with improved renal function in a mouse model of acute kidney injury. Cannabis Cannabinoid Res. 2016, 1, 218–228.
- Li, X.; Liu, Y.; Gong, D.; Hai, K.; Ke, B.; Zuo, Y. The critical role of cannabinoid receptor 2 in URB602-induced protective effects against renal ischemia-reperfusion injury in the rat. Shock 2020, 54, 520–530.
- Zhou, S.; Wu, Q.; Lin, X.; Ling, X.; Miao, J.; Liu, X.; Hu, C.; Zhang, Y.; Jia, N.; Hou, F.F.; et al. Cannabinoid receptor type 2 promotes kidney fibrosis through orchestrating β-catenin signaling. Kidney Int. 2021, 99, 364–381.
- Rothner, A.; Gov, T.; Hinden, L.; Nemirovski, A.; Tam, J.; Rosenzweig, B. Systemic changes in endocannabinoids and endocannabinoid-like molecules in response to partial nephrectomy-induced ischemia in humans. Int. J. Mol. Sci. 2023, 24, 4216.
- McSweeney, K.R.; Gadanec, L.K.; Qaradakhi, T.; Ali, B.A.; Zulli, A.; Apostolopoulos, V. Mechanisms of cisplatin-induced acute kidney injury: Pathological mechanisms, pharmacological interventions, and genetic mitigations. Cancers 2021, 13, 1572.
- Wang, D.; Lippard, S.J. Cellular processing of platinum anticancer drugs. Nat. Rev. Drug Discov. 2005, 4, 307–320.
- Ries, F.; Klastersky, J. Nephrotoxicity induced by cancer chemotherapy with special emphasis on cisplatin toxicity. Am. J. Kidney Dis. 1986, 8, 368–379.
- Zhang, B.; Ramesh, G.; Norbury, C.C.; Reeves, W.B. Cisplatin-induced nephrotoxicity is mediated by tumor necrosis factor-α produced by renal parenchymal cells. Kidney Int. 2007, 72, 37–44.
- Mukhopadhyay, P.; Pan, H.; Rajesh, M.; Bátkai, S.; Patel, V.; Harvey-White, J.; Mukhopadhyay, B.; Haskó, G.; Gao, B.; Mackie, K.; et al. CB1 cannabinoid receptors promote oxidative/nitrosative stress, inflammation and cell death in a murine nephropathy model. Br. J. Pharmacol. 2010, 160, 657–668.
- Pertwee, R.G. Cannabinoid pharmacology: The first 66 years. Br. J. Pharmacol. 2006, 147, S163–S171.
- Castaneto, M.S.; Gorelick, D.A.; Desrosiers, N.A.; Hartman, R.L.; Pirard, S.; Huestis, M.A. Synthetic cannabinoids: Epidemiology, pharmacodynamics, and clinical implications. Drug Alcohol Depend. 2014, 144, 12–41.
- Seely, K.A.; Lapoint, J.; Moran, J.H.; Fattore, L. Spice drugs are more than harmless herbal blends: A review of the pharmacology and toxicology of synthetic cannabinoids. Prog. Neuropsychopharmacol. Biol. Psychiatry 2012, 39, 234–243.
- Wood, K.E. Exposure to bath salts and synthetic tetrahydrocannabinol from 2009 to 2012 in the United States. J. Pediatr. 2013, 163, 213–216.
- Kamijo, Y.; Takai, M.; Fujita, Y.; Sakamoto, T. A multicenter retrospective survey of poisoning after consumption of products containing novel psychoactive substances from 2013 to 2014 in Japan. Am. J. Drug Alcohol Abuse 2016, 42, 513–519.
- Luciano, R.L.; Perazella, M.A. Nephrotoxic effects of designer drugs: Synthetic is not better! Nat. Rev. Nephrol. 2014, 10, 314–324.
- Tait, R.J.; Caldicott, D.; Mountain, D.; Hill, S.L.; Lenton, S. A systematic review of adverse events arising from the use of synthetic cannabinoids and their associated treatment. Clin. Toxicol. 2016, 54, 1–13.
- Alp, A.; Akdam, H.; Avcıoğlu, B.Y.; Ersan, S. Synthetic cannabinoids in the kidneys. Rev. Assoc. Med. Bras. 2017, 63, 10–12.