1. The ECS and Chronic Kidney Disease
1.1. Diabetic Kidney Disease
The function of endocannabinoids and their receptors have been linked to diabetic nephropathy, the leading cause of kidney disease in the United States and an initiator of end stage kidney disease [
5,
54,
55]. Diabetic nephropathy can contribute to a decrease in the life expectancies of patients, and there are currently no fully effective treatments. Multiple studies have been aimed to better elucidate the roles of endocannabinoid signaling and its effects on diabetic nephropathy, highlighting the role endocannabinoid signaling plays in podocyte and renal proximal tubular cell (RPTC) physiology (
Figure 1) [
24,
26,
53].
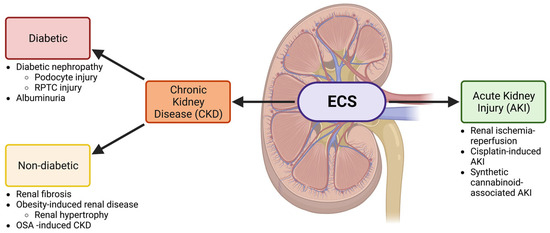
Figure 1. The ECS is involved in both CKD and AKI. A schematic showing the involvement of the ECS in the kidney and the relationship between the nephropathies that will be discussed in this review. Both acute kidney injury (AKI) and chronic kidney disease (CKD) will be investigated. CKD is further divided into diabetic and non-diabetic chronic kidney disease and is associated with several conditions discussed throughout this review. Other abbreviations: renal proximal tubular cell (RPTC) and obstructive sleep apnea (OSA). Created with BioRender.com.
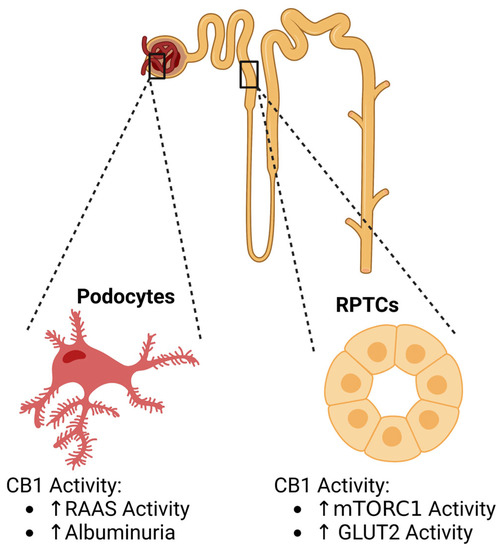
Podocytes are highly specialized cells in the kidney that play important roles in glomerular filtration. Interestingly, CB1 has been identified within the podocytes, allowing the ECS to be a target for study [
53]. It has been well-characterized that the ECS plays a major role in diabetes and metabolic diseases, which in turn brings focus to diabetic nephropathy [
56,
57,
58,
59]. In a 2014 study performed by Jourdan et al., high glucose levels were found to affect CB1 activation in podocytes, causing increased
Cnr1 expression [
54]. The overactivation of CB1 resulted in podocyte damage, due to both hyperglycemia and increased renin–angiotensin–aldosterone (RAAS) activity, causing diabetic nephropathy. The potential therapeutic benefits of blocking endocannabinoid signaling in podocytes and its effect on diabetic nephropathy was studied using JD5037, a peripheral CB1 antagonist. Peripheral CB1 antagonism prevented the deterioration of kidney function when applied to prediabetic mice and helped reverse already seen effects in mice exhibiting diabetic nephropathy. Peripheral CB1 antagonism in Zucker diabetic fatty (ZDF) rats, a popular type-2 diabetes model, prevented hyperglycemia, increased kidney weight, elevated plasma creatine, and increased blood urea nitrogen levels and lead to a reduction in GFR. Xanthine oxidase activity was normalized, and the activation of the RAAS was prevented. Peripheral CB1 antagonism also reversed already developed nephropathy in older mice, including polyuria and albuminuria [
54]. Overall, this study helped to identify the potential pharmaceutical capabilities of CB1 antagonism in the prevention and treatment of diabetic kidney disease.
In addition to peripheral CB1 antagonism, a novel mouse model with a podocyte-specific deletion of CB1 was developed by Jourdan et al. in 2018 [
60]. It has been previously shown that CB1 activation, specifically in the podocytes, contributes to podocyte injury caused by hyperglycemia and increased RAAS activity [
54]. A previous in vivo model highlighted that the addition of high levels of glucose in cultured human podocyte cells created a significant increase in CB1 expression and led to podocyte injury [
58]. The study performed by Jourdan et al. in 2014 did observe a
Cnr1 knockdown model in the presence of high glucose levels to study CB1 blockade [
54]. However, their more recent study used a
Cnr1 deletion model, allowing them to explore the role of CB1 in the development of diabetic nephropathy in vivo. The mice with the podocyte-specific deletion of
Cnr1 were with streptozotocin (STZ) to induce type-1 diabetic nephropathy and chronic hyperglycemia [
60]. The knockout of podocyte-specific
Cnr1 protected glomerular and podocyte function, while also shielding proximal tubular function against injury [
60]. Knockout of
Cnr1 also contributes to a decrease in oxidative stress and inflammation induced by diabetes as compared to controls [
60]. Overall, CB1 has been identified as being a mechanistic component of podocyte injury specifically by influencing oxidative stress [
53,
60].
A specific characteristic of diabetic nephropathy is albuminuria, which results from the dysregulation of the glomerular filtration barrier (
Figure 2) [
61]. CB1 has previously been shown to influence albuminuria, as CB1 blockade was found to ameliorate albuminuria [
57]. In 2018, Barutta et al. investigated how AM6545, a CB1 receptor antagonist, acted in conjunction with the treatment of perindopril, an ACE-inhibitor, for type 1 diabetic mice [
59]. The goal was to see if the connection between ECS and RAS can be applied to a more effective therapy for albuminuria than ACE-inhibitors alone [
59]. Once diabetes was established in their mouse models, albuminuria levels were observed with the inclusion of AM6545, perindopril, or the dual therapy of both. Both the AM6545 and perindopril treatments reduced albuminuria by 50%. However, the dual therapy resulted in an albuminuria level comparable to that of the non-diabetic mice, indicating a rescue early diabetic nephropathy. Additionally, a similar trend in podocyte development was observed in the reduction of nephrin and podocin. The single treatments of perindopril or AM6545 helped to rescue nephrin and podocin reduction in immunostaining; however, dual therapy fully rescued the reduction in diabetic mice. The researchers then looked at whether CB1 receptor interferes with retinoic acid (RA) signaling and found that exposure to RA resulted in an increase in nephrin mRNA level. However, treatment with a CB1 receptor agonist, ACEA, reverted this trend by lowering nephrin expression below the control. Additionally, the rescue of other factors such as monocyte infiltration, macrophage polarization towards M2, or an overabundant expression of inflammatory factors were observed in the dual therapy. This study highlights the therapeutic potential of dual therapies by inhibiting both CB1 and RAS to reverse nephropathy in diabetic mice.
Figure 2. CB1 activity and associations in DKD. Endocannabinoid activity plays roles in podocytes and renal proximal tubule cell (RPTC) physiology. Created with BioRender.com.
Although widely accepted as a glomerular disease, recent studies suggest RPTCs play a role in diabetic kidney disease (DKD) when they are exposed to dysregulated nutrient and energy sensing in diseases such as diabetes and obesity [
62,
63]. Specifically, the activation of mTORC1, due to a nutrient overload, in the RPTCs was identified as a key driver of diabetic nephropathy [
62]. CB1 has been characterized as being expressed within the proximal tubular cells of the human adult kidney and plays roles in RPTC function [
24,
26,
39,
64,
65]. Interestingly, endocannabinoid signaling has been associated with proximal tubular hypertrophy, which is associated with diabetic nephropathy [
64]. As shown by Hinden et al. in 2022 [
63], endocannabinoid signaling plays a key role in regulating mTORC1 signaling in RPTCs in normal and disease states. Hyperglycemia, a known contributing factor of diabetic nephropathy was shown to stimulate endocannabinoid signaling and the subsequent activation of CB1. Therefore, the relationship between CB1 and mTORC1 was studied. CB1-deficient diabetic mice demonstrated a decrease in mTORC1 signaling and levels of GLUT2, preserving kidney function, inferring that CB1 activity stimulates mTORC1 activity, leading to DKD advancement in mice. However, non-diabetic mice that do not contain CB1 show increased mTORC1 signaling, due to increased amino acid transport [
63]. CB1 activity has also been found to mediate obesity-induced renal lipotoxicity by downregulating AMPK signaling and increasing lipid accumulation in the RPTCs. CB1 antagonists may offer therapeutic benefits by preventing GLUT2 translocation to the apical membrane, causing enhanced glucose uptake and increasing AMPK phosphorylation [
63]. Taken together, these studies have revealed crucial physiological changes associated with CB1 in podocytes and RPTCs in diabetes (
Figure 6).
1.2. The ECS in Renal Fibrosis and Other Non-Diabetic CKDs
Chronic kidney disease can also be independent of diabetes. Kidney diseases such as fibrosis and proteinuria can be hereditary or caused by other issues that precede diabetes, such as pregnancy [
66,
67]. Interestingly, the endocannabinoid system has been linked to non-diabetic chronic kidney diseases (NDCKD) as well. Renal fibrosis is caused by the excessive accumulation of an extracellular matrix, and while it may additionally be a result of DKD, it can additionally originate from non-diabetic chronic injury. Interestingly, CB1 has recently emerged as a key player in its pathogenesis (
Figure 3) [
67]. In 2015, Lecru et al. aimed to better understand the underlying mechanisms of renal fibrosis [
29]. In order to do so, they performed a microarray analysis to compare the gene expressions of fibrotic and normal kidneys. Diseased kidneys were studied using the unilateral ureteral obstruction (UUO) experimental model in mice to induce fibrosis. Surprisingly, the Cnr1 gene, the gene responsible for encoding CB1, was one of the most upregulated genes within the UUO model. This novel finding then lead to the exploration of CB1 expression within renal fibrosis and other diseases. The immunostaining of multiple renal biopsies revealed that CB1 expression increased in renal fibrosis, acute interstitial nephritis, IgA nephropathy, and diabetic nephropathy, indicating a relationship between CB1 expression and kidney function. When further studied in the context of renal fibrosis, CB1 expression was found to be increased in myofibroblasts, the main contributor of fibrosis. In the UUO model, global endocannabinoid levels, including AEA and 2-AG, increased, indicating a change in the overall endocannabinoid signaling system within the context of renal fibrosis. To further validate their findings, a genetic knockout (Cnr1
−/−) and pharmacological blockade model were used and showed a decrease in fibrosis. Through these CB1 deficiency models, this study determined that CB1 is involved in the development of renal fibrosis [
29].
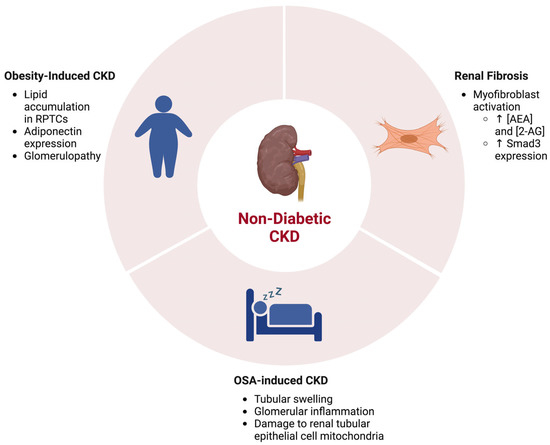
Figure 3. The ECS and associations in NDCKD. Kidney diseases including renal fibrosis, obesity-induced CKD, and obstructive sleep apnea (OSA)-induced CKD are associated with activity alterations in the ECS. Created with BioRender.com.
More recent evidence suggests that CB1 agonism may be influencing renal fibrosis through Smad3 signaling [
68]. Smad3 expression is a major mediator of renal fibrosis [
69,
70]. TGF-β1, a mediator of renal fibrosis, activates Smad3, which in turn regulates target genes for renal fibrosis [
29,
70,
71]. More specifically, aspects of fibrosis such as collogen synthesis and the epithelial–mesenchymal transition are dependent on TGF-β1/Smad3 signaling [
70]. The results from the Lecru et al. study [
29] suggest that CB1 activity acts downstream of TGF-β1. Interestingly, CB1 is hypothesized to be a negative mediator of TGF-β1/Smad3 signaling in pulmonary fibrosis [
72]. The study performed by Golosova et al. [
68] highlighted that high doses of AEA led to an increase in Smad3 expression, however surprisingly did not affect TGF-β1 expression. Previous studies have indicated that CB1 antagonists, such as AM-251, acted upstream of Smad3 and p38 MAPK in EMT; however, it did not act through CB1 [
73]. This finding indicates that CB1 may not play a role in EMT regarding renal fibrosis. It was also hypothesized that CB1 has the potential to act through an alternative pattern, independent of TGF-β1 [
68]. Overall, these findings indicate a complicated role for CB1 signaling in renal fibrosis that needs further exploration.
It has been well characterized that the over-activation of the ECS is associated with obesity and obesity-related issues (
Figure 7) [
74,
75]. As a result, the potential relationship between the ECS and obesity-induced CKD is of interest. A previous study treated obese Zucker rat models with CB1 antagonists, which led to the attenuation of proteinuria and improved creatinine clearance, as well as a reduction in renal hypertrophy [
75]. This connection between renal injury and CB1 activity highlights the role ECS plays in obesity-induced CKD. While less is known about it than obesity-associated glomerulopathy, renal tubule injury is another facet of obesity-induced CKD that is a potential therapeutic target [
76]. A study, performed by Udi et al. in 2017 [
65], aimed to focus on RPTCs within the context of obesity-induced CKD, as the RPTCs are particularly sensitive to lipid accumulation. It has been previously shown that the ECS is a key player in obesity; it has been shown to be involved in RPTC function as well, highlighting a possible relationship between endocannabinoid signaling and obesity-related renal pathologies [
64,
74,
75]. Using RPTC-specific CB1-null mice, CB1 deletion was determined to reduce obesity-induced lipid accumulation in the kidney, inferring CB1 plays a role in the development of renal lipotoxicity. This result was then associated with the regulation of the LKB1/AMPK/ACC signaling pathway by the CB1R-coupled Gai/o-PKA axis [
65]. Further studies indicated that CB1 helps to regulate the expression of adiponectin, an enzyme involved in glucose regulation and fatty acid degradation [
77]. Adiponectin is also regulated by induced NOS (iNOS), another pathway involved in renal dysfunction, which allowed researchers to explore the connection between CB1 signaling and iNOS activity. The association of these two signaling pathways led to the establishment of a CB1/iNOS hybrid antagonist as a possible treatment for obesity-induced CKD [
77].
Another NDCKD that has been studied in the context of the ECS is obstructive sleep apnea (OSA)-induced chronic kidney disease (
Figure 7). The renal issues associated with OSA are mainly caused by intermittent hypoxia, more specifically [
78]. Chronic intermittent hypoxia (CIH) has previously been shown to activate CB1 within bone, triggering metabolic bone disorders, indicating a relationship between CB1 and CIH [
79]. It was hypothesized that this disease is due to mitochondrial dysfunctions that cause organ damage and might be able to be regulated by endocannabinoids. CIH is one of the main symptoms of OSA and affects kidney mitochondria. Therefore, patients with untreated OSA can result in CKD [
80]. CB1 has been shown to affect mitochondrial function in kidneys and is shown to be very active in human kidney disease [
24]. Zhao et al. in 2021 [
81] therefore looked at three mitochondrial regulators as well as CB1 expression in the OSA-CIH rat model after the treatment of the CB1 antagonist, rimonabant (Ri). Through the analysis of renal tissue under electron microscopy (EM) and western blot analysis, no morphological abnormalities were found in the control group; however, the CIH group had swelling and a narrow tubule lumen. Tubular epithelial cells were also damaged, and a slightly inflammation was seen within the glomeruli. With the rimonabant treatment, swelling and tubular damage decreased. The CIH group indicated severe structural damage in renal tubular epithelial cell mitochondria, as well as the presence of mitochondria fragments. After rimonabant treatment, the damage to the mitochondria, as well as the presence of mitochondrial fragments, diminished, suggesting that the antagonist helped combat some of the structural damage in the renal mitochondria due to the CIH. Western blotting showed that CB1 expression in the renal tissues of the CIH group was increased, and after treatment with the antagonist, CB1 levels decreased, subsequently decreasing renal injury. Overall, it was concluded that CIH caused by OSA could induce endocannabinoid signaling, which then correlates to renal disease.
2. The ECS and Acute Kidney Injury
Renal ischemia-reperfusion (IR) has been known to contribute to AKI development. IR is caused by the impairment and rescue of blood flow to the kidney, commonly associated with injuries associated with partial nephrectomy (PN), transplant, infarction, and sepsis [
82,
83]. The endocannabinoid system (ECS) has been studied in its relationship with the IR across organs [
84]. In the case of IR, the involvement of ECS has been observed in rodent models. For example, it has been observed that renal clamping causes changes in the levels of AEA [
85] or 2-AG [
43,
86]. Additionally, the expression of CB1 and CB2 have also been found to be changed [
85,
87]. In Rothner et al., 2023 [
88], the researchers studied the systemic endocannabinoid (eCB) levels caused by surgical renal IR. The study looked at 16 patients undergoing on-clamp PN and studied several levels of several factors important to the ECS system before renal ischemia, after 10 min of ischemia, and after 10 min from blood reperfusion. The study found that levels of 2-AG and kidney dysfunction biomarkers positively correlated with each other. The study showed that the levels of BUN, glucose, and sCr were elevated after reperfusion. The study did not detect significant changes in the CBs, CB-like molecules, or arachidonic acid (AA) in all patients. The study also showed that there was a significant increase in
N-acylethanolamines in non-obese patients, however not in obese patients who already had higher levels of
N-acylethanolamines.
Another issue that currently involves AKI is the inclusion of cisplatin-induced AKI (CIAKI). Cisplatin is a well-established chemotherapeutic agent that can result in AKI in its patients [
89,
90,
91]. This chemotherapeutic drug induces tumor necrosis factor-α (TNF-α), which in turn stimulates cytokine and chemokine expression within the kidney to cause injury [
92]. Although this drug is widely used to treat a plethora of cancers, a treatment to reduce the incidences of AKI under its use remains unknown. A study by Mukhopadhyay et al., 2010 [
50], aimed to begin exploring a possible connection between CIAKI and the role CB1 plays in oxidative stress and inflammation. In this study, the researchers investigated the effect of pharmacological and genetic inhibition of CB1 receptors on a murine nephropathy model. First, they detected AEA and 2-AG in the mouse kidney and found that cisplatin increased the tissue levels of AEA. Additionally, it was found that with the treatment of AM-281 and SR141716, two CB1 antagonists, renal dysfunction was attenuated. A similar trend was additionally observed in CB1
−/− mice. In addition, the researchers looked at histopathological damages and cell death and found that both the pharmacological and genetic inhibition of CB1 helped attenuate the renal damage, as well as reducing cell death in murine kidneys. The researchers further investigated additional factors of stress, such as p38 and JNK MAPK activation or interrelated oxidative/nitrosative stress and found that both the genetic deletion and pharmacological inhibition of CB1 helped mitigate these markers of renal dysfunction. Taken together, these results suggested that CB1 inhibitors could serve as a powerful pharmacotherapy treatment to reduce the nephrotoxicity effects of cisplatin.
Synthetic cannabinoids (SCs) are increasingly known as an abusive drug in young adults. Initially developed in the 1960s for studies on the pharmacology of cannabinoid receptors, SCs are non-polar agonists of the two endocannabinoid receptors, CB1 and CB2 [
93,
94]. However, not much has been known about their specific pharmacology [
95]. SCs have recently become a popular type of drug abuse in young adults, particularly due to the similarities in their effects to cannabis [
96]. Additionally, SCs are easily accessible with a low cost and distributed under names such as “Spice” or “K2”. In addition, they are easily consumed, either by smoking, insufflating, or ingesting. The most common renal pathology for renal problems due to SCs are acute kidney injury (AKI), accounting for less than 1% of all cases [
97,
98,
99]. SCs have been known for their ability to reduce inotropic effects, leading to a blood flow reduction to the kidney. SC use can also be associated with cannabinoid hyperemesis syndrome, a condition observed in long-term users who experience symptoms such as vomiting, abdominal discomfort, or nausea [
100].
This entry is adapted from the peer-reviewed paper 10.3390/cells12101419