Major histocompatibility complex class I (MHC I) plays a crucial role in the development of adaptive immune response in vertebrates. MHC molecules are cell surface protein complexes loaded with short peptides and recognized by the T-cell receptors (TCR). Peptides associated with MHC are named immunopeptidome. The MHC I immunopeptidome is produced by the proteasome degradation of intracellular proteins. The knowledge of the immunopeptidome repertoire facilitates the creation of personalized antitumor or antiviral vaccines. A huge number of publications on the immunopeptidome diversity of different human and mouse biological samples - plasma, peripheral blood mononuclear cells (PBMCs), and solid tissues, including tumors - appeared in the scientific journals in the last decade. Significant immunopeptidome identification efficiency was achieved by advances in technology: the immunoprecipitation of MHC and mass spectrometry-based approaches.
- immunopeptidome
- major histocompatibility complex class I
- MHC
- human leukocyte antigen
- HLA
- immunoaffinity chromatography
1. Introduction
The studies on transplantation in the 20th century led to the discovery of the antigens determining the compatibility of various tissues during transplantation [1]. These antigens were found to be presented by special transmembrane protein complexes called major histocompatibility complexes (MHCs). In humans, the products of this gene family were first found on leukocytes. Hence, the genes were called human leukocyte antigen (HLA) genes [2]. There are four groups of HLA genes (classes I, II, III, and IV), which are all located on chromosome 6. The products of these genes are proteins that differ in structure and function [3]. The HLA I and HLA II genes are among the most polymorphic human genes, and as of October 2020, 28,786 different alleles have been described for them (https://www.ebi.ac.uk/ipd/imgt/hla/stats.html). HLA I genes include the most common HLA-A, HLA-B, and HLA-C, and rare HLA-E, HLA-F, and HLA-G genes. HLA II genes incorporate HLA-DRA, HLA-DRB, HLA-DQA, HLA-DQB, HLA-DPA, HLA-DPB, HLA-DMA, HLA-DMB, HLA-DOA, and HLA-DOB [4]. The products of expression of these genes are transmembrane glycoproteins that present peptide antigens on the cell surface. The exceptions are HLA-DMA, HLA-DMB, HLA-DOA, and HLA-DOB, which regulate the loading of peptides onto MHC II molecules [5]. The main function of these molecules is to participate in the T cell-mediated immune response.
Genome-wide association studies (GWAS) have demonstrated a strong association between the presence of certain diseases and a specific HLA genotype [6,7,8,9,10]. Moreover, in several cases, the cause is HLA single nucleotide polymorphism, which affects the binding of the peptide antigen to HLA and thereby alters the repertoire of antigens presented to T cells. For example, in GWAS of 2000 Parkinson’s disease (PD) cases and 1986 healthy donors, a strong association was found between the risk of PD and the expression of the HLA-DRB5*01 and HLA-DRB1*15:01 alleles [9]. These alleles exist in about one-third of PD patients. At least one epitope obtained during the degradation of α-synuclein, which forms insoluble fibrils in filamentous inclusions of Lewy bodies in PD, is specifically presented by these forms of HLA II [11]. Similar studies made it possible to associate systemic sclerosis with the expression of HLA-DRB1*15∶02 and HLA-DRB1*16∶02 (585 cases and 458 controls) [12] and psoriasis with HLA-C*06:02 (461 psoriatic patients and 454 healthy controls) [13]. According to in silico analysis of the binding affinity of each possible fragment of the SARS-CoV-2 proteins with the expression products of 145 HLA-A, HLA-B, and HLA-C alleles, the protein product of the HLA-B*46:01 allele had the fewest predicted binding of SARS-CoV-2 peptides, which indicates a more severe course of coronavirus infection in carriers of this allele. On the contrary, the product of the HLA-B*15:03 allele is more capable of presenting highly conserved peptides of SARS-CoV-2, which improves the capabilities of T cell-based immunity [14]. Interestingly, persistent expression of a particular HLA gene can simultaneously lower the risk of developing one disease and increase the risk of developing another. For example, the instability of HLA-C makes the body more susceptible to HIV, which is why the virus seeks to suppress the expression of this gene using the Vpu protein [15,16]. On the other hand, with increased expression of HLA-C, the occurrence of Crohn’s disease [17] and psoriatic arthritis [13,18] becomes more likely.
The development of mass spectrometry and peptidomic approaches to the isolation and identification of low-presented native peptides made it possible to directly determine the MHC ligands. As part of personalized cancer therapy development, mass spectrometry-based immunopeptidomics has gained the interest of biotechnological and pharmaceutical companies in the determination of peptide antigens for clinical application [34]. The goal of cancer immunotherapy is to activate the patient’s immune system and recruit their T cells, especially the CD8+ T cells, to fight the tumor. Complexes of HLA I molecules with antigenic peptides are the key to activate T killer cells. There are a significant number of oncoimmunotherapy approaches: the utilization of checkpoint blockade [35], chimeric antigen receptor (CAR) T-cell therapy [36], T-cell receptor (TCR)-engineered cells [37], T cell adoptive cell transfer (ACT) [38], and oncolytic viruses (OV)-based immunotherapy [39]. Identification of the tumor-specific immunopeptidome, as well as strategies for the isolation and genetic modification of T cells, are essential in the development of personalized cancer immunotherapy [40,41]. The diverse repertoire of HLA I presented on tumor cells is a good source of potential tumor antigens [42]. In 2018, Hilf et al. published a trial of novel personalized therapeutic vaccines (APVAC1 and APVAC2) for glioblastoma as part of the glioma actively personalized vaccine consortium (GAPVAC) [43]. The creation of these vaccines utilized published technology that includes the search for immunogenic neoantigens based on transcriptome and immunopeptidome analysis of the patient’s tumor tissue [44]. The immunogenicity of the identified peptides was verified using CD8+ T cells isolated from the patient’s blood. This highly personalized form of immunotherapy was first implemented in a global project involving a large number of research studies from various scientific centers.
2. Methods for Immunopeptidome Analysis
An important milestone in the studies of the immunopeptidome of various animal cells was a creation of the method for the isolation of MHC I ligands by mild acid elution (MAE) proposed by Sugawara et al. in 1987 [68]. The essence of this easy-to-implement method is the short-term treatment of living cells with citrate buffer (pH 3.0). As a result of such treatment, the β2 microglobulin molecule non-covalently bound to the MHC I heavy chain dissociates, destabilizing the structure of the entire complex. This reduces the peptide-binding capacity of the HLA-A, HLA-B, and HLA-C complexes, i.e., it leads to the loss of peptides associated with the MHC class I molecules [68]. The hypothesis was made that MHC class II molecules do not lose their antigens during MAE, which increases the specificity of the technique. The assumption was confirmed a little later [69]. Importantly, working with cells by the MAE method leaves them viable with the ability to regenerate MHC I complexes with antigens, which facilitates the accumulation of a significant amount of MHC I ligands. At the time MAE was proposed, which allowed using no more than 100 million cells, it was indeed an extremely effective technique compared to other methods used for the isolation of the MHC I peptidome (trifluoroacetic acid extraction [70] and immunoaffinity isolation using specific antibodies [65]), requiring 1–10 billion cells. The growing interest in immunopeptidomics and a significant amount of accumulated experimental data have stimulated the emergence of several detailed reviews and comparative works related to the MAE method [71,72,73,74,75]. Undoubtedly, the simplicity and efficiency of MAE [68], including a small number of purification steps, the absence of detergents [72], the possibility of multiple processing of living cells [76], and the reduction of losses in the case of working with low-affinity peptides [72] made the MAE method one of the main tools of immunopeptidomics. On the other hand, the need to work with living cells is one of the most significant weaknesses of the MAE method, which is highlighted by many researchers. In addition, elution should take place in a cell suspension; that is, cells should circulate freely in solution [73]. Hence, it is not possible to use MAE on tissues and cell lines requiring special conditions for growth. Even more problematic is the simultaneous elution of peptides present in large amounts on the cell surface and not related to the MHC I ligandome. According to Fortier et al., only about 40% of all peptides isolated by the MAE method are associated with MHC class I, while the rest are contaminants [68,72,77].
Immunoaffinity chromatography is a method for the isolation and purification of a target substance from a multicomponent mixture based on a specific non-covalent interaction of an antibody immobilized on a solid support and an antigenic epitope of the target substance [78]. Unlike MAE, immunoaffinity chromatography finds applications in various fields of biomedicine, including clinical diagnostics, detection of substances hazardous to the environment, and pharmacological research [79]. The basic principle of immunity chromatography is still the same, despite the constant improvement of methodology [74,80,81,82]. A multicomponent mixture featuring a cell line lysate, homogenized tissue, or biological fluid sample is incubated with MHC-specific antibodies pre-immobilized on magnetic particles or agarose-based polymeric resins as solid support (Figure 1) [79]. The murine monoclonal antibody, clone W6/32, which specifically binds to the α2–α3 heavy chain region of the products of all classical genes HLA-A, HLA-B, and HLA-C is commonly used [74,82]. After purification from non-specifically bound substances, MHC molecules together with associated peptides are eluted. Currently, the method of immunoaffinity purification is the most commonly used for isolating an immunopeptidome. There are reasons for this: (1) most of the peptides isolated by this method can be true ligands of MHC; several studies bioinformatically confirm the high affinity for MHC in about 90% of identifications [83,84,85], and (2) this method is less demanding on the biomaterial; it is possible to use both cell lines and tissues, biological fluids, including frozen samples.
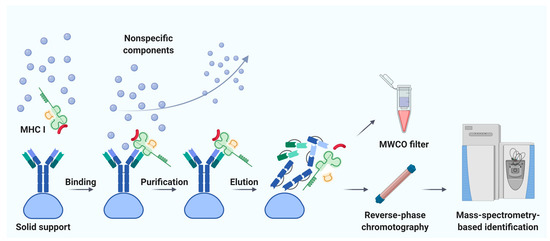
Figure 1. Immunoaffinity chromatography. Abbreviations: MHC I, major histocompatibility complex class I; MWCO, molecular weight cutoff. Created with BioRender.com.
Noteworthy, the labor and time costs of this method are higher compared to MAE. Immunoaffinity chromatography for the isolation of MHC requires a significant amount of specific antibodies; therefore, there is a need to maintain an in-house hybridoma producing the required antibodies [86,87]. On average, about 1 mg of antibodies per sample is required [88]. It is not surprising that, to our knowledge, the largest published work to date is devoted to the study of the immunopeptidome of only 10 biological samples of postoperative material and 142 samples of blood plasma [89]. Using isotopically labeled peptides, Hassan and co-authors found that losses during immunoprecipitation of the MHC ligandome reached 90–99% [90]. Due to the large number of washes required to get rid of non-specific peptides, there is a high risk of losing low-affinity MHC ligands [71]. In addition, it is still not precisely established how universal the antibodies are—that is, whether there are such MHC variants that bind antibodies with low affinity and, as a result, some of the MHC-ligand complexes are lost [91]. Taking into account all sources of loss, it is not surprising that the number of cells required for successful LC-MS/MS identification of the MHC ligandome varies from 100 million to 10 billion [92]. However, attempts are being made to improve methods of immunoaffinity purification [93]. Chong et al. propose to accelerate and automate the protocol by carrying out immunoprecipitation in 96-well plates. The researchers isolated 42,556 unique MHC class I associated peptides belonging to 8975 precursor proteins, using 21 wells containing 100 million cells each [93]. Out of 10 million cells, they managed to identify only 1846 peptides, but these 1846 peptides are almost the same as the most represented peptides isolated from 100 million cells. Lanoix and co-authors published a comparison of the quality of the B-cell lymphoblast immunopeptidome isolation by MAE and immunoprecipitation [73]. As a result of the isolation of immunopeptidome from 2, 20, and 100 million cells, the authors managed to identify 2016, 3931, and 5093 unique peptides by immunoaffinity chromatography and 314, 2081, and 2996 unique peptides by MAE with MS detection. Thus, more peptides associated with HLA I were obtained by immunoaffinity purification. However, the difference in the total amount of isolated peptides with an increase in the initial number of cells aligns between the two methods.
It is the isolation of the immunopeptidome that some authors aptly call an Achilles’ heel, hinting at an inhibitory effect on the development of the research area as a whole [88]. Indeed, back in 1992, Hunt et al. showed that the majority of peptides presented via MHC I varies from 100 to 1000 copies per cell, and only a few are present in 1000 to 3000 molecules per cell [80]. In some cases, the representation of a single peptide can reach 10,000 copies per cell [94]. Moreover, according to the data of Schuster et al., the average number of HLA I molecules per cell varies from 5000 to 150,000 [95], and according to Lanoix et al., the total number of MHC I per cell can reach 0.5–3 million [73], which theoretically allows the cell to present 10,000–30,000 different peptides. If we take into account that losses during immunoprecipitation of the MHC I ligandome can reach 90–99% [90], we can isolate 1 to 300 million molecules of each peptide from 1 million cells, which approximately corresponds to amounts from 2 amol to 0.5 fmol. As the limiting sensitivity of LC-MS/MS, one can take the result obtained by Matthias Mann’s group in 2010 on Orbitrap Exactive [96]. Using the Universal Proteomics Standard (UPS1), they identified 348 different peptides, in triplicate, from 45 of 48 UPS1 proteins using the 140 fmol of corresponding tryptic peptides. Although the identification was performed against a database of all human proteins, the sensitivity would be lower under conditions of a high dynamic range of real biological samples. If we take 500 fmol of a peptide as a sufficient amount, then for successful identification of the peptide in the immunopeptidome, at least 1 billion cells should be taken, which is roughly consistent with the scale of current works on immunoprecipitation [88,90,92,95].
The study on the regulation of the presentation of the HLA I peptide repertoire is an important task [50,97,98,99]. The detection of factors capable of increasing the amount of MHC presented by a cell can reduce the required volume of biological material and/or increase the number of different detectable MHC ligands. Javitt and co-authors show that pro-inflammatory cytokines tumor necrosis factor alpha (TNFα) and interferon gamma (IFNγ) increase the number of identifiable HLA I ligands in the lung epithelial cell line A549 from 3444 unique peptides without cytokine treatment to 6582 unique peptides after the treatment [99]. About 500 million cells were used in a single experiment. The authors showed that the pro-inflammatory molecules TNFα and INFγ increased the diversity of immunopeptidome, which was due to the functioning of a special immunoproteasome synthesized in cells under the effect of these cytokines [49].
Another method for isolation of HLA I molecules and their ligandome is the transfection of a cell line with an expression vector encoding a soluble secreted form of MHC I, without a transmembrane domain, and the further immunoprecipitation of secreted MHCs with peptides attached. The MHC I delivery methods include DNA transfection [100,101], transduction with retroviruses [102], and mRNA transfection [103]. At the same time, this method allows culturing cells for long periods, similar to MAE, which facilitates the accumulation of a significant amount of MHC ligands and gives the most specific result due to the immunoprecipitation. However, various genetic engineering procedures can cause an appreciable rearrangement of the protein composition of the cell, together with the MHC ligandome. In addition, similar to MAE, this method does not work with tissues due to the complexity of the use of genetic engineering techniques [74].
3. Future Perspectives
Over the past 20–25 years, research in the field of immunopeptidomics has made significant progress, both in methodological terms and in the volume and completeness of the data obtained. The accumulated experience made it possible to improve the immunotherapeutic approach to various oncological diseases [162]. Immunopeptidome analysis has become one of the essential directions of working with adaptive immunity. However, the application of various methodological approaches to this analysis has led to the results that not only cannot be compared with each other but also combined into large associative studies. In the largest to date, to our knowledge, the immunopeptidome study analyzed 10 biosamples of postoperative material and 142 blood plasma samples taken from patients with glioblastoma [89]. Therefore, without a single unified protocol for working with an immunopeptidome, the scientific community faces the difficulty of comparing and combining data and gaining even greater knowledge about the immunopeptidome.
To unite the efforts of the community of immunopeptidome researchers under the auspices of the Human Proteome Organization (HUPO), the Human Immunopeptidome Project (HIPP) consortium was created [163]. The main stated objective of the HUPO-HIPP is to map the entire repertoire of HLA ligands and to make immunopeptidome analysis accessible to any researcher. In particular, this project seeks to conduct association studies of the immunopeptidome in a consortium of large centers or even countries, which could provide a global qualitative and comprehensive analysis of various disease-associated HLA alleles. The idea of such large-scale studies has led Vizcaíno et al. to the concept of an immunopeptidome-wide association study (IWAS): combining the capabilities of many scientific centers to identify correlations between the components of an immunopeptidome and certain human diseases based on studies of large groups of people [164].
We assume that further optimization of research in the field of immunopeptidome will be the automation of experiments to reduce time costs and increase reproducibility. High-throughput protocols for the isolation of immunopeptidome are already beginning to appear, which can allow the robotization of this process [93]. Similar to the massive parallel nucleic acid sequencing, similar strategies for parallel peptide sequencing are already being developed [165], which will allow the analysis of low-abundance samples and mapping of rare amino acid variants.
This entry is adapted from the peer-reviewed paper 10.3390/molecules25225409