This entry is focused on recent studies about the mutual interactions of key mediators of AhR and Wnt/β-catenin signaling pathways and on the assessment of the complexity of the crosstalk between the AhR signaling cascade and the canonical Wnt pathway. AhR performs many endogenous functions by integrating its signaling pathway into organ homeostasis and into the maintenance of crucial cellular functions and biological processes. The Wnt signaling pathway regulates cell proliferation, differentiation, and many other phenomena, and this regulation is important for embryonic development and the dynamic balance of adult tissues. AhR and Wnt are the main signaling pathways participating in the control of cell fate and function and occupy a central position in a variety of processes linked with development and various pathological conditions.
- AhR
- Wnt
- β-catenin
- Mutual interaction
- Crosstalk
1. AhR and AhR Signaling
Aryl hydrocarbon receptor (AhR) is a ligand-activated transcription factor [1][2][3]. Initially, AhR was identified in studies on its binding to polychlorinated aromatic hydrocarbons [1][2][3]. AhR is ubiquitously expressed in many vertebrate cell types and mediates various cellular functions [4][5][6]. AhR regulates fundamental metabolic processes that modulate such cellular phenomena as proliferation, differentiation, the cell cycle, adhesion, migration, invasion, pluripotency, and stemness and mediate such physiological functions of AhR as the homeostasis of organs of the cardiac, immune, reproductive, nervous, and intestinal systems and of the liver, insulin–glucose regulation, oxidative stress, and adaptation to hypoxia [7][8][9]. It is believed that the evolution of AhR, which began hundreds of millions of years ago in multicellular animals, has been driven by adaptation to environmental toxins Nonetheless, the following hypothesis is likely to be true: the function of AhR was originally the regulation of development, and its involvement in xenobiotic metabolism probably has emerged as an adaptation later, during the evolution of vertebrates [4][10].
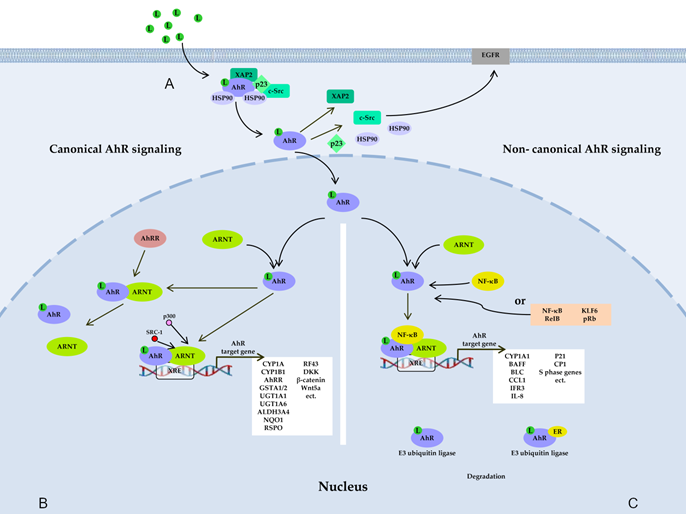
Figure 1. AhR signaling pathways. (A) Before ligand binding, AhR is located in the cytosol in the form of a complex with hepatitis B virus X-associated protein 2 (XAP-2), with two heat shock proteins 90 (HSP90), with cochaperone p23, and with additional partners, including c-Src. A ligand changes AhR’s conformation, thereby leading to the dissociation of the complex and translocation of AhR to the nucleus, where AhR forms a dimer either with AhR nuclear transporter (ARNT) (canonical pathway) (B) or with partner proteins other than ARNT (noncanonical pathway) (C), such as transcription factor Krüppel-like factor 6 (KLF6), transcription factors of the nuclear factor kappa B (NF-κB) family, retinoblastoma protein (pRb), or nuclear receptors (e.g., estrogen receptor α). The resultant dimer binds to a xenobiotic-responsive element (XRE) in DNA and, thus, induces the transcription of target genes of AhR. AhR also takes part in nongenomic signaling: when dissociated from the c-Src complex, AhR can interact with epidermal growth factor receptor (EGFR), whose downstream signaling includes the focal adhesion kinase FAK pathway and mitogen-activated protein kinase (MAPK) pathways called RAS–RAF–MEK1/2–ERK1/2 and AKT–PI3K–mTOR, as well as protein kinase C (PKC), STAT proteins, SRC, and NF-κB. AhR is also a component of a protein complex that functions as an E3 ubiquitin ligase.
2. Wnt and Wnt Signaling
The signaling pathway of the Wnt (wingless-type MMTV integration site) family of secreted glycolipoproteins is central to multiple developmental processes [11]. Compared to the signaling pathways of other growth factors, Wnt signaling has several unique properties and, in fact, governs development and tissue homeostasis. The Wnt signaling pathway takes part in the workings of such crucial cellular phenomena as intercellular communication, cell migration, the determination of the fate of nascent cells, embryogenesis, organogenesis, and homeostasis of embryonic and adult stem cells [11][12][13]. The Wnt signaling pathway is evolutionarily conserved and Wnt proteins are active in all clades of the animal kingdom [14].
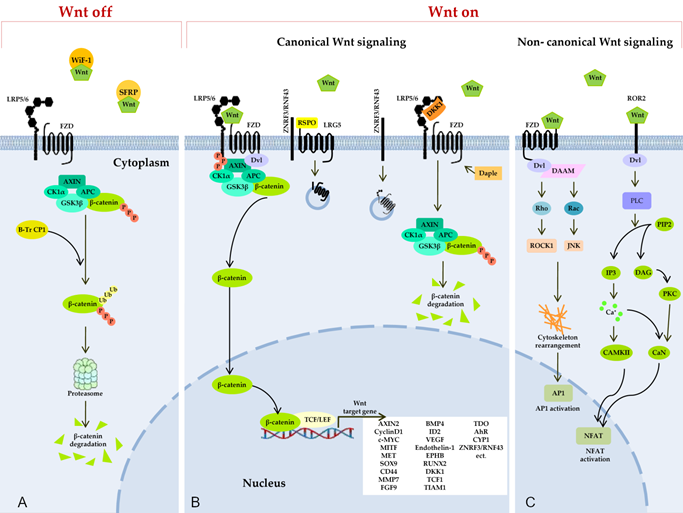
Figure 2. Wnt signaling pathways. (A) Secreted Frizzled-related proteins (SFRPs) and Wnt-inhibitory factor 1 (WIF1) counteract Wnt signaling by isolating the Wnt protein in the extracellular space. In the absence of Wnt proteins, degradation of β-catenin takes place in the cytoplasm, and this process limits the transcription of Wnt target genes. The degradation of cytosolic β-catenin is mediated by proteasomes or autophagy because of persistent phosphorylation of β-catenin by a multimeric protein degradation complex containing Axin, adenomatous polyposis colonic (APC) scaffold proteins, casein kinase 1α (CK1α), and glycogen synthase kinase 3 (GSK3) and because of subsequent ubiquitination by β-transducin repeat-containing protein 1 (β-TrCP1). (B) Binding of a Wnt protein (usually Wnt3A and Wnt1) to a receptor called Frizzled (FZD) and to coreceptor LRP5/6 induces phosphorylation of the cytoplasmic region of LRP5/6 by GSK3β and recruitment of a cytosolic protein called Disheveled (Dvl). This event leads to an increase in the amount of β-catenin in the cytoplasm, owing to isolation and inhibition of the protein complex responsible for β-catenin phosphorylation. After translocation to the nucleus, β-catenin forms an active complex with transcription factor T-cell factor and a transcription factor from the lymphoid enhancer-binding factor family (TCF/LEF) and other histone-modifying coactivators (CBP, p300, and BCL9) and initiates the transcription of target genes of Wnt. The Daple protein may inhibit the recruitment of DVL. A secreted inhibitor by the name of Dickkopf (DKK) can also counteract Wnt signal transduction by competitively binding to LRP5/6. Ubiquitin ligases ZNRF3/RNF43 reduce the availability of membrane receptors (LRP5/6) and FZD to Wnt through their internalization and degradation, thereby negatively regulating the Wnt pathway. R-spondin (RSPO) binds to receptor proteins called leucine rich repeat-containing G protein-coupled receptor 4, 5, or 6 (LGR4/5/6) and to the extracellular domain of RNF43/ZNRF3, resulting in their physical association, internalization into the cytoplasm, and the removal of ZNRF3/RNF43 from the membrane. In this way, the availability of the Wnt membrane receptor increases, and the activation of the Wnt ligand–mediated pathway is enhanced. (C) Binding of Wnt isoforms (e.g., Wnt-5a) to either FZD or Ror2 triggers noncanonical (β-catenin–independent) Wnt signaling cascades, including the inhibition of the canonical Wnt/β-catenin pathway. In the planar cell polarity (PCP) pathway, Wnt binds to receptors FZD, thereby activating Dvl and Dvl-associated activator of morphogenesis (DAAM). Dvl and DAAM together trigger small GTPases Rho and Rac, which catalyze the activation of JNK and ROCK1, resulting in a cytoskeleton rearrangement and changes in gene expression via activation of the activating protein-1 (AP1) family of transcription factors. In the Wnt/Ca2+ pathway, a signal is transmitted via a triggering of phospholipase C (PLC) through Dvl activation. PLC hydrolyzes phosphatidylinositol 4,5-bisphosphate (PIP2) generating inositol 1,4,5-triphosphate (IP3) and 1,2-diacylglycerol (DAG). IP3 activates calcium/calmodulin-dependent protein kinase II (CAMKII) and calcineurin (CaN) through a release of calcium from the endoplasmic reticulum. Active CAMKII and CaN alter a target gene’s expression by inducing a transcription factor called nuclear factor of activated T cells (NFAT). DAG in turn triggers protein kinase C, which raises CaN activity.
3. Intersection of AhR and Wnt Signaling
Between AhR and the Wnt/β-catenin pathways, various types of crosstalk can occur, which have been most fully described in a review by Schneider et al. [15]. In recent years, numerous studies showed potential crosstalk between AhR and Wnt/β-catenin signaling cascades, and a lot of new evidence was accumulated about a dual interaction between these pathways, as did data on the interdependent regulation of their signal transduction as a possible mechanism for the maintenance of physiological or pathophysiological functions.
It can be concluded that AhR has been identified as an inhibitor of canonical Wnt signaling when a specific role has been tested in projects using only a knockout or knockdown of AhR. For instance, a loss of AhR in murine CD11c+ intestinal cells is associated with aberrant Wnt signaling caused by overexpression of target genes Axin2, Lgr5, с-Myc, Nmp, and Dkk3 in intestinal macrophages and anomalous development of the intestinal epithelium [16]. In intestinal stem cells, AhR modulates the expression of ubiquitin ligases RNF43 and ZNRF3 negatively regulating Wnt/β-catenin signaling and thus limiting cell hyperproliferation, as shown in an AhR−/− mouse model and cultured intestinal organoids [17]. A knockout of AhR enhances Wnt signal transduction in the mouse colon epithelium [18]. The AhR−/− mouse liver phenotype includes faster cell proliferation with insufficient cell polyploidization and activation of Wnt/β-catenin signaling, possibly owing to AhR being a part of a repressive complex promoting β-catenin ubiquitination and its subsequent degradation [19]. At least coimmunoprecipitation of AhR and β-catenin has been registered under normal conditions in the adult liver [19]. AhR deficiency improves liver regeneration after acute toxic damage by CCl4 or after severe mechanical damage to the organ but contributes to the development of hepatocarcinoma, probably in part due to the activation of β-catenin, which is one of pluripotency factors [20]. A knockdown of AhR in murine Schwann cell line MSC80 launches the Wnt/β-catenin pathway, by raising the protein level of active β-catenin and mRNA expression of signaling components Lrp6, Dvl2, Dvl3, and Axin2, by activating TCF/LEF-binding sites [21]. Only one study indicates that AhR signal transduction upregulates the expression of β-catenin and Wnt5a/b, where a knockdown of the AhR gene led to suppression of Wnt5a expression [22]. This was documented in tumor tissue from patients with inflammatory breast cancer demonstrating constitutive overexpression of AhR.
AhR has been identified as an inhibitor or activator of the canonical Wnt signaling pathway when AhR’s role has been determined in various studies by means of its agonistic ligands such as environmental pollutants (TCDD, PM2.5, 3MC, or BaP), an organophosphate pesticide (CPF), natural compounds (I3C and AS-IV), or endogenous compounds (FICZ, IS, or Kyn). In some reports, such AhR agonists as TCDD, PM2.5, FICZ, IS, Kyn, and I3C have been shown to impede Wnt/β-catenin signaling [23][24][25][26][27][28][29][30][31][32][33][34][35]. Such data have been obtained in research on zebrafish embryogenesis [26], on the heart of zebrafish embryos[27], on mouse embryonic stem cells [24], on rat liver progenitors [23], on human undifferentiated HepaRG liver progenitors [28], on the P19 cell line, which is a malignant analog of embryonic stem cells [29], on the mouse urogenital sinus [24][25], on lung carcinoma A549 cells [36], on mesenchymal stem cells from mice with collagen-induced arthritis [30], on human myofibroblasts [31], on intestinal epithelial organoids of C57BL/6 mice [33], on human NIH 3T3 fibroblasts, human kidney HK-2 epithelial cells, and mesenchymal embryonic fibroblasts [34], on colon carcinoma HT-29 cells [35], and on mouse hippocampal neuronal HT22 cells [32].
Wnt/β-catenin pathway signals in turn are involved in interactions with AhR signaling. An analysis of recent research findings shows that the Wnt/β-catenin pathway is mainly a positive regulator of the expression of AhR and of its target genes in various cell types [37][38][39][40][41]. β-Catenin plays the main part in the interaction with AhR signaling. Firstly, AhR and CYP1 have been identified as target genes of β-catenin [37][42]; secondly, activated β-catenin may elevate the amount of AhR [37][43][44]; thirdly, the binding sites for β-catenin/TCF and AhR in the promoter of CYP1A are located in close proximity [40][45]; and finally, β-catenin interacts with a XRE as a coactivator of the AhR–ARNT complex [37][38][39]. Lately, in chronic myeloid leukemia cell lines, a regulatory interaction between AhR and the Dvl protein, which plays a central role in β-catenin stabilization, has been demonstrated; this interaction manifests itself as a dependence of AhR protein expression on the silencing or overexpression of Dvl [41]. There is an example of a different influence on AhR signaling in HCT116 human colon carcinoma cells, where the suppression of β-catenin potentiated the induction of the expression/activity of CYP1 enzymes by BaP [46]. It is worth highlighting the involvement of key enzymes of the Kyn pathway of tryptophan metabolism—TDO2 and IDO1 (which catalyze the formation of Kyn, an agonist of AhR)—in the interactions between AhR and Wnt. Enhancement of the AhR and β-catenin pathways can proceed in a coordinated manner and is a consequence of activation of the IDO1 enzyme: after triggering of IDO1 and subsequent activation of AhR, β-catenin is activated [47]. The activation of AhR mediated by TDO2 enhances the expression of target genes of Wnt/β-catenin: CCND1, RNF43, ZRNF3, LGR5, and ASCL2 [48].
Taken together, the presented literature proves the important role of the interdependent regulation of AhR and Wnt/β-catenin signaling pathways in the maintenance of embryogenesis, organogenesis, and cell homeostasis under normal and pathophysiological conditions. In these interactions of the signaling pathways, AhR appears to act as a fine-tuner of Wnt signal transduction in a cellular, tissue, or organ context; this property means high potential of AhR as a therapeutic target for Wnt pathway modulation in pathological conditions, including cancer and nonmalignant diseases.
This entry is adapted from the peer-reviewed paper 10.3390/cimb45050248
References
- Hankinson O; The aryl hydrocarbon receptor complex. Annu Rev Pharmacol Toxicol 1995, 35, 307-340, 10.1146/annurev.pa.35.040195.001515.
- Hankinson O.; Role of coactivators in transcriptional activation by the aryl hydrocarbon receptor. Arch Biochem Biophys 2005, 433(2), 379-386, 10.1016/j.abb.2004.09.031.
- Yin J, Sheng B, Qiu Y, Yang K, Xiao W, Yang H.; Role of AhR in positive regulation of cell proliferation and survival. Cell Prolif 2016, 49(5), 554-560, 10.1111/cpr.12282.
- Sondermann NC, Faßbender S, Hartung F, et al.; Functions of the aryl hydrocarbon receptor (AHR) beyond the canonical AHR/ARNT signaling pathway. Biochem Pharmacol 2023, 208, 115371, 10.1016/j.bcp.2022.115371.
- Safe S, Han H, Goldsby J, Mohankumar K, Chapkin RS.; Aryl Hydrocarbon Receptor (AhR) Ligands as Selective AhR Modulators: Genomic Studies. Curr Opin Toxicol 2018, 11-12, 10-20, 10.1016/j.cotox.2018.11.005.
- Hahn ME.; Aryl hydrocarbon receptors: diversity and evolution. Chem Biol Interact 2002, 141(1-2), 131-160, 10.1016/s0009-2797(02)00070-4.
- Roman ÁC, Carvajal-Gonzalez JM, Merino JM, Mulero-Navarro S, Fernández-Salguero PM.; The aryl hydrocarbon receptor in the crossroad of signalling networks with therapeutic value. Pharmacol Ther 2018, 185, 50-63, 10.1016/j.pharmthera.2017.12.003.
- Sayed TS, Maayah ZH, Zeidan HA, Agouni A, Korashy HM.; Insight into the physiological and pathological roles of the aryl hydrocarbon receptor pathway in glucose homeostasis, insulin resistance, and diabetes development. Cell Mol Biol Lett 2022, 27(1), 103, 10.1186/s11658-022-00397-7.
- Zablon HA, Ko CI, Puga A.; Converging Roles of the Aryl Hydrocarbon Receptor in Early Embryonic Development, Maintenance of Stemness, and Tissue Repair. Toxicol Sci 2021, 182(1):, 1-9, 10.1093/toxsci/kfab050.
- Hahn ME, Karchner SI, Merson RR.; Diversity as Opportunity: Insights from 600 Million Years of AHR Evolution. Curr Opin Toxicol 2017, 2, 58-71, 10.1016/j.cotox.2017.02.003.
- Liu J, Xiao Q, Xiao J, et al.; Wnt/β-catenin signalling: function, biological mechanisms, and therapeutic opportunities. Signal Transduct Target Ther 2022, 7(1), 3, 10.1038/s41392-021-00762-6.
- Jung YS, Park JI.; Wnt signaling in cancer: therapeutic targeting of Wnt signaling beyond β-catenin and the destruction complex. Exp Mol Med 2020, 52(2), 183-191, 1038/s12276-020-0380-6.
- Lorzadeh S, Kohan L, Ghavami S, Azarpira N.; Autophagy and the Wnt signaling pathway: A focus on Wnt/beta-catenin signaling. Biochim Biophys Acta Mol Cell Res 2021, 1868(3), 118926, 10.1016/j.bbamcr.2020.118926.
- Loh KM, van Amerongen R, Nusse R.; Generating Cellular Diversity and Spatial Form: Wnt Signaling and the Evolution of Multicellular Animals. Dev Cell 2016, 38(6), 643-655, 10.1016/j.devcel.2016.08.011.
- Schneider AJ, Branam AM, Peterson RE.; Intersection of AHR and Wnt signaling in development, health, and disease. Int J Mol Sci 2014, 15(10), 17852-17885, 10.3390/ijms151017852.
- Chng SH, Kundu P, Dominguez-Brauer C, et al.; Ablating the aryl hydrocarbon receptor (AhR) in CD11c+ cells perturbs intestinal epithelium development and intestinal immunity. Sci Rep 2016, 6, 23820, 10.1038/srep23820.
- Metidji A, Omenetti S, Crotta S, et al.; The Environmental Sensor AHR Protects from Inflammatory Damage by Maintaining Intestinal Stem Cell Homeostasis and Barrier Integrity. Immunity 2019, 50(6), 1542, 10.1016/j.immuni.2019.05.024.
- Han H, Davidson LA, Hensel M, et al.; Loss of Aryl Hydrocarbon Receptor Promotes Colon Tumorigenesis in ApcS580/+; KrasG12D/+ Mice. Mol Cancer Res 2021, 19(5), 771-783, 10.1158/1541-7786.MCR-20-0789.
- Moreno-Marín N, Merino JM, Alvarez-Barrientos A, et al.; Aryl Hydrocarbon Receptor Promotes Liver Polyploidization and Inhibits PI3K, ERK, and Wnt/β-Catenin Signaling. iScience 2018, 4, 44-63, 10.1016/j.isci.2018.05.006.
- Rejano-Gordillo CM, González-Rico FJ, Marín-Díaz B, et al.; Liver regeneration after partial hepatectomy is improved in the absence of aryl hydrocarbon receptor. Sci Rep 2022, 12(1), 15446, 10.1038/s41598-022-19733-0.
- Shackleford G, Sampathkumar NK, Hichor M, et al.; Involvement of Aryl hydrocarbon receptor in myelination and in human nerve sheath tumorigenesis. Proc Natl Acad Sci U S A 2018, 115(6), E1319-E1328, 10.1073/pnas.1715999115.
- Mohamed HT, Gadalla R, El-Husseiny N, et al.; Inflammatory breast cancer: Activation of the aryl hydrocarbon receptor and its target CYP1B1 correlates closely with Wnt5a/b-β-catenin signalling, the stem cell phenotype and disease progression. J Adv Res 2018, 16, 75-86, 10.1016/j.jare.2018.11.006.
- Faust D, Vondráček J, Krčmář P, et al.; AhR-mediated changes in global gene expression in rat liver progenitor cells. Arch Toxicol 2013, 87(4), 681-698, 10.1007/s00204-012-0979-z.
- Wang Q, Kurita H, Carreira V, et al.; Ah Receptor Activation by Dioxin Disrupts Activin, BMP, and WNT Signals During the Early Differentiation of Mouse Embryonic Stem Cells and Inhibits Cardiomyocyte Functions. Toxicol Sci 2016, 149(2), 346-357, 10.1093/toxsci/kfv246.
- Branam AM, Davis NM, Moore RW, Schneider AJ, Vezina CM, Peterson RE.; TCDD inhibition of canonical Wnt signaling disrupts prostatic bud formation in mouse urogenital sinus. Toxicol Sci 2013, 133(1), 42-53, 10.1093/toxsci/kft027.
- Wincent E, Stegeman JJ, Jönsson ME. 2015; 284(2):163-179. doi:10.1016/j.taap.2015.02.014; Combination effects of AHR agonists and Wnt/β-catenin modulators in zebrafish embryos: Implications for physiological and toxicological AHR functions. Toxicol Appl Pharmacol 2015, 284(2), 163-179, 10.1016/j.taap.2015.02.014.
- Zhang M, Chen J, Jiang Y, Chen T.; Fine particulate matter induces heart defects via AHR/ROS-mediated endoplasmic reticulum stress. Chemosphere 2022, 307(Pt 2), 135962, 10.1016/j.chemosphere.2022.135962.
- Svobodová J, Procházková J, Kabátková M, et al.; 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) Disrupts Control of Cell Proliferation and Apoptosis in a Human Model of Adult Liver Progenitors. Toxicol Sci 2019, 172(2), 368-384, 10.1093/toxsci/kfz202.
- Chen T, Jin H, Wang H, et al.; Aryl hydrocarbon receptor mediates the cardiac developmental toxicity of EOM from PM2.5 in P19 embryonic carcinoma cells. Chemosphere 2019, 216, 372-378, 10.1016/j.chemosphere.2018.10.160.
- Tong Y, Niu M, Du Y, et al.; Aryl hydrocarbon receptor suppresses the osteogenesis of mesenchymal stem cells in collagen-induced arthritic mice through the inhibition of β-catenin. Exp Cell Res 2017, 350(2), 349-357, 10.1016/j.yexcr.2016.12.009.
- Woeller CF, Roztocil E, Hammond CL, Feldon SE, Phipps RP.; The Aryl Hydrocarbon Receptor and Its Ligands Inhibit Myofibroblast Formation and Activation: Implications for Thyroid Eye Disease. Am J Pathol 2016, 186(12), 3189-3202, 10.1016/j.ajpath.2016.08.017.
- Duan Z, Zhang S, Liang H, et al.; Amyloid β neurotoxicity is IDO1-Kyn-AhR dependent and blocked by IDO1 inhibitor . Signal Transduct Target Ther 2020, 5(1), 96, 10.1038/s41392-020-0188-9.
- Park JH, Choi AJ, Kim SJ, Cheong SW, Jeong SY.; AhR activation by 6-formylindolo[3,2-b]carbazole and 2,3,7,8-tetrachlorodibenzo-p-dioxin inhibit the development of mouse intestinal epithelial cells. Environ Toxicol Pharmacol 2016, 43, 44-53, 10.1016/j.etap.2016.02.007.
- Arinze NV, Yin W, Lotfollahzadeh S, et al.; Tryptophan metabolites suppress the Wnt pathway and promote adverse limb events in chronic kidney disease. J Clin Invest 2022, 132(1), e142260, 10.1172/JCI142260.
- Park JH, Lee JM, Lee EJ, Kim DJ, Hwang WB.; Kynurenine promotes the goblet cell differentiation of HT-29 colon carcinoma cells by modulating Wnt, Notch and AhR signals. Oncol Rep 2018, 39(4), 1930-1938, 10.3892/or.2018.6266.
- Procházková J, Strapáčová S, Svržková L, et al.; Adaptive changes in global gene expression profile of lung carcinoma A549 cells acutely exposed to distinct types of AhR ligands. Toxicol Lett 2018, 292, 162-174, 10.1016/j.toxlet.2018.04.024.
- Braeuning A, Köhle C, Buchmann A, Schwarz M.; Coordinate regulation of cytochrome P450 1a1 expression in mouse liver by the aryl hydrocarbon receptor and the beta-catenin pathway. Toxicol Sci 2011, 122(1), 16-25, 10.1093/toxsci/kfr080.
- Mulholland DJ, Dedhar S, Coetzee GA, Nelson CC.; Interaction of nuclear receptors with the Wnt/beta-catenin/Tcf signaling axis: Wnt you like to know?. Endocr Rev 2005, 26(7), 898-915, 10.1210/er.2003-0034.
- Shiizaki K, Kido K, Mizuta Y.; Insight into the relationship between aryl-hydrocarbon receptor and β-catenin in human colon cancer cells. PLoS One 2019, 14(11), e0224613, 10.1371/journal.pone.0224613.
- Vaas S, Kreft L, Schwarz M, Braeuning A.; Cooperation of structurally different aryl hydrocarbon receptor agonists and β-catenin in the regulation of CYP1A expression. Toxicology 2014, 325, 31-41, 10.1016/j.tox.2014.08.010.
- Caliskan C, Yuce Z, Ogun Sercan H.; Dvl proteins regulate SMAD1, AHR, mTOR, BRD7 protein expression while differentially regulating canonical and non-canonical Wnt signaling pathways in CML cell lines. Gene 2023, 854, 147109, 10.1016/j.gene.2022.147109.
- Loeppen S, Koehle C, Buchmann A, Schwarz M.; A beta-catenin-dependent pathway regulates expression of cytochrome P450 isoforms in mouse liver tumors. Carcinogenesis 2005, 26(1), 239-248, 10.1093/carcin/bgh298.
- Braeuning A, Sanna R, Huelsken J, Schwarz M.; Inducibility of drug-metabolizing enzymes by xenobiotics in mice with liver-specific knockout of Ctnnb1. Drug Metab Dispos 2009, 37(5), 1138-1145, 10.1124/dmd.108.026179.
- Kasai S, Ishigaki T, Takumi R, Kamimura T, Kikuchi H.; Beta-catenin signaling induces CYP1A1 expression by disrupting adherens junctions in Caco-2 human colon carcinoma cells. Biochim Biophys Acta 2013, 1830(3), 2509-2516, 10.1016/j.bbagen.2012.11.007.
- Schulthess P, Löffler A, Vetter S, et al.; Signal integration by the CYP1A1 promoter--a quantitative study. Nucleic Acids Res 2015, 43(11), 5318-5330, 10.1093/nar/gkv423.
- Kabátková M, Zapletal O, Tylichová Z, et al.; Inhibition of β-catenin signalling promotes DNA damage elicited by benzo[a]pyrene in a model of human colon cancer cells via CYP1 deregulation. Mutagenesis 2015, 30(4), 565-576, 10.1093/mutage/gev019.
- Chen CT, Wu PH, Hu CC, et al.; Aberrant Upregulation of Indoleamine 2,3-Dioxygenase 1 Promotes Proliferation and Metastasis of Hepatocellular Carcinoma Cells via Coordinated Activation of AhR and β-Catenin Signaling. Int J Mol Sci 2021, 22(21), 11661, 10.3390/ijms222111661.
- Miyazaki T, Chung S, Sakai H, et al.; Stemness and immune evasion conferred by the TDO2-AHR pathway are associated with liver metastasis of colon cancer. Cancer Sci 2022, 113(1), 170-181, 10.1111/cas.15182.
- Kasai S, Ishigaki T, Takumi R, Kamimura T, Kikuchi H.; Beta-catenin signaling induces CYP1A1 expression by disrupting adherens junctions in Caco-2 human colon carcinoma cells. Biochim Biophys Acta 2013, 1830(3), 2509-2516, 10.1016/j.bbagen.2012.11.007.
- Schulthess P, Löffler A, Vetter S, et al.; Signal integration by the CYP1A1 promoter--a quantitative study. Nucleic Acids Res 2015, 43(11), 5318-5330, 10.1093/nar/gkv423.
- Kabátková M, Zapletal O, Tylichová Z, et al.; Inhibition of β-catenin signalling promotes DNA damage elicited by benzo[a]pyrene in a model of human colon cancer cells via CYP1 deregulation. Mutagenesis 2015, 30(4), 565-576, 10.1093/mutage/gev019.
- Chen CT, Wu PH, Hu CC, et al.; Aberrant Upregulation of Indoleamine 2,3-Dioxygenase 1 Promotes Proliferation and Metastasis of Hepatocellular Carcinoma Cells via Coordinated Activation of AhR and β-Catenin Signaling. Int J Mol Sci 2021, 22(21), 11661, 10.3390/ijms222111661.
- Miyazaki T, Chung S, Sakai H, et al.; Stemness and immune evasion conferred by the TDO2-AHR pathway are associated with liver metastasis of colon cancer. Cancer Sci 2022, 113(1), 170-181, 10.1111/cas.15182.
- Chng SH, Kundu P, Dominguez-Brauer C, et al.; Ablating the aryl hydrocarbon receptor (AhR) in CD11c+ cells perturbs intestinal epithelium development and intestinal immunity. Sci Rep 2016, 6, 23820, 10.1038/srep23820.