Dilated cardiomyopathy (DCM) represents one of the most common causes of non-ischemic heart failure, characterised by ventricular dilation alongside systolic dysfunction. Despite advances in therapy, DCM mortality rates remain high, and it is one of the leading causes of heart transplantation. Developments in complementary diagnostic procedures, namely cardiac magnetic resonance and genetic testing, have shed new light on DCM understanding and management.
- dilated cardiomyopathy
- risk stratification
- cardiovascular magnetic resonance
1. Introduction
2. Diagnostic Workup
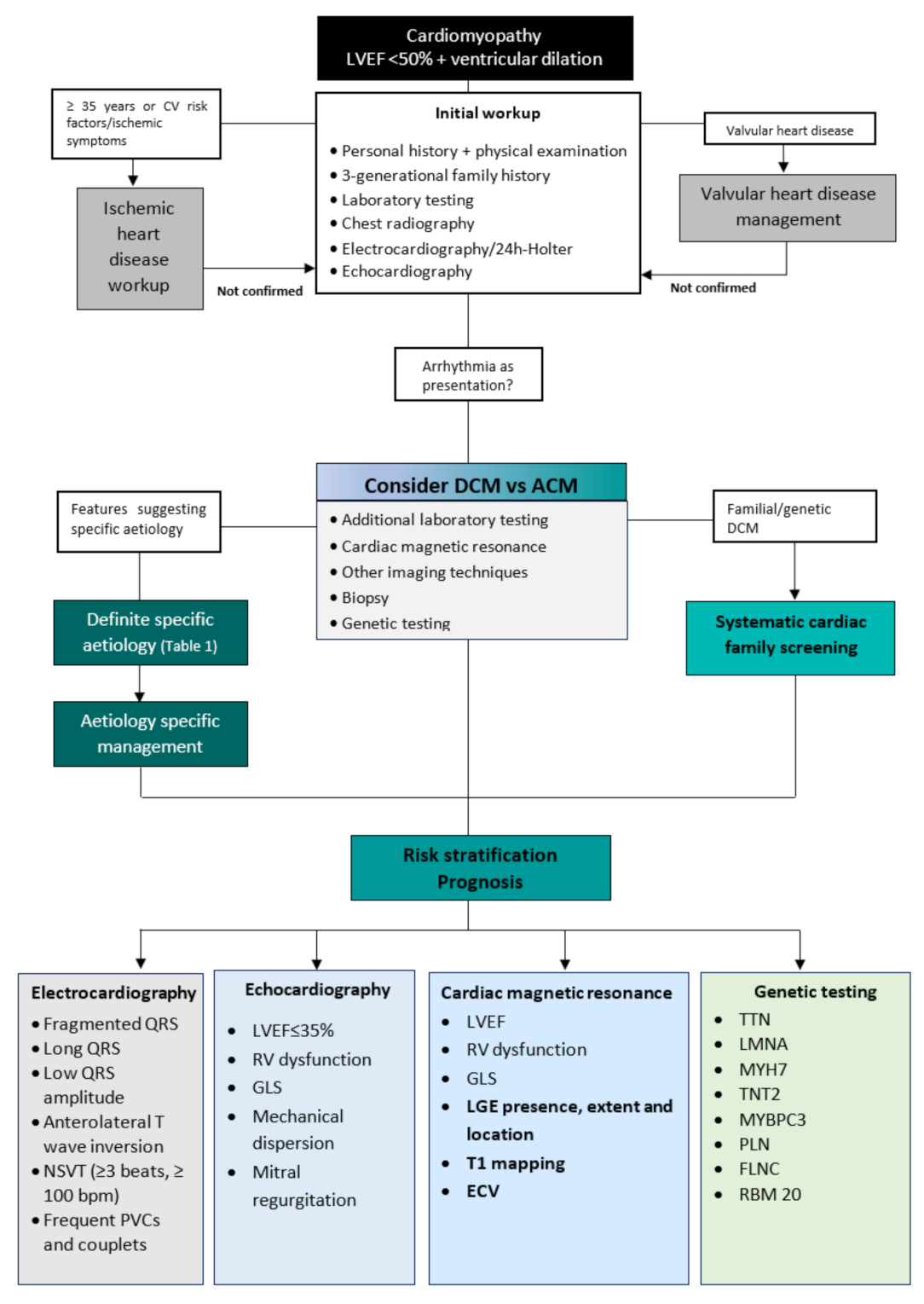
3. Differential Diagnosis
Differential Diagnosis of Dilated and Arrhythmogenic Cardiomyopathy
4. Aetiologies
DCM is a heterogeneous disease encompassing various underlying causes, including genetic and acquired disorders. A positive family history can be detected in up to 30–50% of DCM cases, and a causative genetic mutation can be identified in up to 40% of DCM cases [2][6][26]. In the presence of positive cases in the family, sarcomeric, neuromuscular, and mitochondrial disorders are the most frequent aetiologies. External factors such as exposure to toxins, diabetes, arrhythmia, myocarditis, and pregnancy often contribute to the development of the phenotype and outcome [26]. The non-genetic causes of DCM include infectious (viral or non-viral), autoimmune, toxic, infiltrative-related causes, nutritional deficiencies, and endocrine disorders [26].
5. Risk Stratification and Prognosis
5.1. Electrocardiogram
5.2. Echocardiography
5.3. Cardiac Magnetic Resonance
5.3.1. Late Gadolinium Enhancement
5.3.2. T1 and Extracellular Volume
5.3.3. Feature-Tracking Strain Analysis
5.4. Genetic Testing
6. Prediction of Left Ventricle Reverse Remodelling
This entry is adapted from the peer-reviewed paper 10.3390/biomedicines11030834
References
- Pinto, Y.M.; Elliott, P.M.; Arbustini, E.; Adler, Y.; Anastasakis, A.; Böhm, M.; Duboc, D.; Gimeno, J.; de Groote, P.; Imazio, M.; et al. Proposal for a revised definition of dilated cardiomyopathy, hypokinetic non-dilated cardiomyopathy, and its implications for clinical practice: A position statement of the ESC working group on myocardial and pericardial diseases. Eur. Heart J. 2016, 37, 1850–1858.
- Hershberger, R.E.; Hedges, D.J.; Morales, A. Dilated cardiomyopathy: The complexity of a diverse genetic architecture. Nat. Rev. Cardiol. 2013, 10, 531–547.
- Lund, L.H.; Edwards, L.B.; Dipchand, A.I.; Goldfarb, S.; Kucheryavaya, A.Y.; Levvey, B.J.; Meiser, B.; Rossano, J.W.; Yusen, R.D.; Stehlik, J. The Registry of the International Society for Heart and Lung Transplantation: Thirty-third Adult Heart Transplantation Report-2016; Focus Theme: Primary Diagnostic Indications for Transplant. J. Heart Lung Transplant. 2016, 35, 1158–1169.
- Halliday, B.P.; Gulati, A.; Ali, A.; Newsome, S.; Lota, A.; Tayal, U.; Vassiliou, V.S.; Arzanauskaite, M.; Izgi, C.; Krishnathasan, K.; et al. Sex- and age-based differences in the natural history and outcome of dilated cardiomyopathy. Eur. J. Heart Fail. 2018, 20, 1392–1400.
- Donal, E.; Delgado, V.; Bucciarelli-Ducci, C.; Galli, E.; Haugaa, K.H.; Charron, P.; Voigt, J.U.; Cardim, N.; Masci, P.G.; Galderisi, M.; et al. Multimodality imaging in the diagnosis, risk stratification, and management of patients with dilated cardiomyopathies: An expert consensus document from the European Association of Cardiovascular Imaging. Eur. Heart J. Cardiovasc. Imaging 2019, 20, 1075–1093.
- Japp, A.G.; Gulati, A.; Cook, S.A.; Cowie, M.R.; Prasad, S.K. The Diagnosis and Evaluation of Dilated Cardiomyopathy. J. Am. Coll. Cardiol. 2016, 67, 2996–3010.
- Rapezzi, C.; Arbustini, E.; Caforio, A.L.P.; Charron, P.; Gimeno-Blanes, J.; Heliö, T.; Linhart, A.; Mogensen, J.; Pinto, Y.; Ristic, A.; et al. Diagnostic work-up in cardiomyopathies: Bridging the gap between clinical phenotypes and final diagnosis. A position statement from the ESC Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 2012, 34, 1448–1458.
- Grimm, W.; Christ, M.; Bach, J.; Müller, H.H.; Maisch, B. Noninvasive arrhythmia risk stratification in idiopathic dilated cardiomyopathy: Results of the Marburg Cardiomyopathy Study. Circulation 2003, 108, 2883–2891.
- Patel, A.R.; Kramer, C.M. Role of Cardiac Magnetic Resonance in the Diagnosis and Prognosis of Nonischemic Cardiomyopathy. JACC Cardiovasc. Imaging 2017, 10, 1180–1193.
- Guigui, S.A.; Horvath, S.A.; Arenas, I.A.; Mihos, C.G. Cardiac geometry, function and mechanics in left ventricular non-compaction cardiomyopathy with preserved ejection fraction. J. Echocardiogr. 2022, 20, 144–150.
- Srivastava, S.; Yavari, M.; Al-Abcha, A.; Banga, S.; Abela, G. Ventricular non-compaction review. Heart Fail. Rev. 2022, 27, 1063–1076.
- Regitz-Zagrosek, V.; Roos-Hesselink, J.W.; Bauersachs, J.; Blomström-Lundqvist, C.; Cífková, R.; De Bonis, M.; Iung, B.; Johnson, M.R.; Kintscher, U.; Kranke, P.; et al. 2018 ESC Guidelines for the management of cardiovascular diseases during pregnancy. Eur. Heart J. 2018, 39, 3165–3241.
- Davis, M.B.; Arany, Z.; McNamara, D.M.; Goland, S.; Elkayam, U. Peripartum Cardiomyopathy: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2020, 75, 207–221.
- Hershberger, R.E.; Givertz, M.M.; Ho, C.Y.; Judge, D.P.; Kantor, P.F.; McBride, K.L.; Morales, A.; Taylor, M.R.G.; Vatta, M.; Ware, S.M. Genetic evaluation of cardiomyopathy: A clinical practice resource of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2018, 20, 899–909.
- Miles, C.; Finocchiaro, G.; Papadakis, M.; Gray, B.; Westaby, J.; Ensam, B.; Basu, J.; Parry-Williams, G.; Papatheodorou, E.; Paterson, C.; et al. Sudden Death and Left Ventricular Involvement in Arrhythmogenic Cardiomyopathy. Circulation 2019, 139, 1786–1797.
- Corrado, D.; Perazzolo Marra, M.; Zorzi, A.; Beffagna, G.; Cipriani, A.; Lazzari, M.; Migliore, F.; Pilichou, K.; Rampazzo, A.; Rigato, I.; et al. Diagnosis of arrhythmogenic cardiomyopathy: The Padua criteria. Int. J. Cardiol. 2020, 319, 106–114.
- Spezzacatene, A.; Sinagra, G.; Merlo, M.; Barbati, G.; Graw, S.L.; Brun, F.; Slavov, D.; Di Lenarda, A.; Salcedo, E.E.; Towbin, J.A.; et al. Arrhythmogenic Phenotype in Dilated Cardiomyopathy: Natural History and Predictors of Life-Threatening Arrhythmias. J. Am. Heart Assoc. 2015, 4, e002149.
- Corrado, D.; van Tintelen, P.J.; McKenna, W.J.; Hauer, R.N.W.; Anastastakis, A.; Asimaki, A.; Basso, C.; Bauce, B.; Brunckhorst, C.; Bucciarelli-Ducci, C.; et al. Arrhythmogenic right ventricular cardiomyopathy: Evaluation of the current diagnostic criteria and differential diagnosis. Eur. Heart J. 2020, 41, 1414–1429.
- Towbin, J.A.; McKenna, W.J.; Abrams, D.J.; Ackerman, M.J.; Calkins, H.; Darrieux, F.C.C.; Daubert, J.P.; de Chillou, C.; DePasquale, E.C.; Desai, M.Y.; et al. 2019 HRS expert consensus statement on evaluation, risk stratification, and management of arrhythmogenic cardiomyopathy. Heart Rhythm. 2019, 16, e301–e372.
- Hoorntje, E.T.; Te Rijdt, W.P.; James, C.A.; Pilichou, K.; Basso, C.; Judge, D.P.; Bezzina, C.R.; van Tintelen, J.P. Arrhythmogenic cardiomyopathy: Pathology, genetics, and concepts in pathogenesis. Cardiovasc. Res. 2017, 113, 1521–1531.
- Chun, K.H.; Oh, J.; Hong, Y.J.; Yu, H.T.; Lee, C.J.; Kim, T.H.; Joung, B.; Pak, H.N.; Lee, M.H.; Kim, Y.J.; et al. Prognostic Cardiac Magnetic Resonance Markers of Left Ventricular Involvement in Arrhythmogenic Cardiomyopathy for Predicting Heart Failure Outcomes. J. Am. Heart Assoc. 2022, 11, e023167.
- Cipriani, A.; Bauce, B.; De Lazzari, M.; Rigato, I.; Bariani, R.; Meneghin, S.; Pilichou, K.; Motta, R.; Aliberti, C.; Thiene, G.; et al. Arrhythmogenic Right Ventricular Cardiomyopathy: Characterization of Left Ventricular Phenotype and Differential Diagnosis With Dilated Cardiomyopathy. J. Am. Heart Assoc. 2020, 9, e014628.
- Segura-Rodríguez, D.; Bermúdez-Jiménez, F.J.; Carriel, V.; López-Fernández, S.; González-Molina, M.; Oyonarte Ramírez, J.M.; Fernández-Navarro, L.; García-Roa, M.D.; Cabrerizo, E.M.; Durand-Herrera, D.; et al. Myocardial fibrosis in arrhythmogenic cardiomyopathy: A genotype-phenotype correlation study. Eur. Heart J. Cardiovasc. Imaging 2020, 21, 378–386.
- Cipriani, A.; Mattesi, G.; Bariani, R.; Cecere, A.; Martini, N.; De Michieli, L.; Da Pozzo, S.; Corradin, S.; De Conti, G.; Zorzi, A.; et al. Cardiac magnetic resonance imaging of arrhythmogenic cardiomyopathy: Evolving diagnostic perspectives. Eur. Radiol. 2023, 33, 270–282.
- Groeneweg, J.A.; van der Zwaag, P.A.; Olde Nordkamp, L.R.; Bikker, H.; Jongbloed, J.D.; Jongbloed, R.; Wiesfeld, A.C.; Cox, M.G.; van der Heijden, J.F.; Atsma, D.E.; et al. Arrhythmogenic right ventricular dysplasia/cardiomyopathy according to revised 2010 task force criteria with inclusion of non-desmosomal phospholamban mutation carriers. Am. J. Cardiol. 2013, 112, 1197–1206.
- Bondue, A.; Arbustini, E.; Bianco, A.; Ciccarelli, M.; Dawson, D.; De Rosa, M.; Hamdani, N.; Hilfiker-Kleiner, D.; Meder, B.; Leite-Moreira, A.F.; et al. Complex roads from genotype to phenotype in dilated cardiomyopathy: Scientific update from the Working Group of Myocardial Function of the European Society of Cardiology. Cardiovasc. Res. 2018, 114, 1287–1303.
- Goldberger, J.J.; Subačius, H.; Patel, T.; Cunnane, R.; Kadish, A.H. Sudden cardiac death risk stratification in patients with nonischemic dilated cardiomyopathy. J. Am. Coll. Cardiol. 2014, 63, 1879–1889.
- Marume, K.; Noguchi, T.; Tateishi, E.; Morita, Y.; Kamakura, T.; Ishibashi, K.; Noda, T.; Miura, H.; Nishimura, K.; Nakai, M.; et al. Mortality and Sudden Cardiac Death Risk Stratification Using the Noninvasive Combination of Wide QRS Duration and Late Gadolinium Enhancement in Idiopathic Dilated Cardiomyopathy. Circ. Arrhythmia Electrophysiol. 2018, 11, e006233.
- Merlo, M.; Cannatà, A.; Gobbo, M.; Stolfo, D.; Elliott, P.M.; Sinagra, G. Evolving concepts in dilated cardiomyopathy. Eur. J. Heart Fail. 2018, 20, 228–239.
- Daubert, J.P.; Zareba, W.; Hall, W.J.; Schuger, C.; Corsello, A.; Leon, A.R.; Andrews, M.L.; McNitt, S.; Huang, D.T.; Moss, A.J. Predictive value of ventricular arrhythmia inducibility for subsequent ventricular tachycardia or ventricular fibrillation in Multicenter Automatic Defibrillator Implantation Trial (MADIT) II patients. J. Am. Coll. Cardiol. 2006, 47, 98–107.
- Jenkins, C.; Bricknell, K.; Hanekom, L.; Marwick, T.H. Reproducibility and accuracy of echocardiographic measurements of left ventricular parameters using real-time three-dimensional echocardiography. J. Am. Coll. Cardiol. 2004, 44, 878–886.
- Becker, M.A.J.; Cornel, J.H.; van de Ven, P.M.; van Rossum, A.C.; Allaart, C.P.; Germans, T. The Prognostic Value of Late Gadolinium-Enhanced Cardiac Magnetic Resonance Imaging in Nonischemic Dilated Cardiomyopathy: A Review and Meta-Analysis. JACC Cardiovasc. Imaging 2018, 11, 1274–1284.
- Di Marco, A.; Anguera, I.; Schmitt, M.; Klem, I.; Neilan, T.G.; White, J.A.; Sramko, M.; Masci, P.G.; Barison, A.; McKenna, P.; et al. Late Gadolinium Enhancement and the Risk for Ventricular Arrhythmias or Sudden Death in Dilated Cardiomyopathy: Systematic Review and Meta-Analysis. JACC Heart Fail. 2017, 5, 28–38.
- Halliday, B.P.; Baksi, A.J.; Gulati, A.; Ali, A.; Newsome, S.; Izgi, C.; Arzanauskaite, M.; Lota, A.; Tayal, U.; Vassiliou, V.S.; et al. Outcome in Dilated Cardiomyopathy Related to the Extent, Location, and Pattern of Late Gadolinium Enhancement. JACC Cardiovasc. Imaging 2019, 12, 1645–1655.
- aus dem Siepen, F.; Buss, S.J.; Messroghli, D.; Andre, F.; Lossnitzer, D.; Seitz, S.; Keller, M.; Schnabel, P.A.; Giannitsis, E.; Korosoglou, G.; et al. T1 mapping in dilated cardiomyopathy with cardiac magnetic resonance: Quantification of diffuse myocardial fibrosis and comparison with endomyocardial biopsy. Eur. Heart J. Cardiovasc. Imaging 2015, 16, 210–216.
- Puntmann, V.O.; Carr-White, G.; Jabbour, A.; Yu, C.Y.; Gebker, R.; Kelle, S.; Hinojar, R.; Doltra, A.; Varma, N.; Child, N.; et al. T1-Mapping and Outcome in Nonischemic Cardiomyopathy: All-Cause Mortality and Heart Failure. JACC Cardiovasc. Imaging 2016, 9, 40–50.
- Chen, Z.; Sohal, M.; Voigt, T.; Sammut, E.; Tobon-Gomez, C.; Child, N.; Jackson, T.; Shetty, A.; Bostock, J.; Cooklin, M.; et al. Myocardial tissue characterization by cardiac magnetic resonance imaging using T1 mapping predicts ventricular arrhythmia in ischemic and non-ischemic cardiomyopathy patients with implantable cardioverter-defibrillators. Heart Rhythm 2015, 12, 792–801.
- Vita, T.; Gräni, C.; Abbasi, S.A.; Neilan, T.G.; Rowin, E.; Kaneko, K.; Coelho-Filho, O.; Watanabe, E.; Mongeon, F.P.; Farhad, H.; et al. Comparing CMR Mapping Methods and Myocardial Patterns Toward Heart Failure Outcomes in Nonischemic Dilated Cardiomyopathy. JACC Cardiovasc. Imaging 2019, 12, 1659–1669.
- Romano, S.; Judd, R.M.; Kim, R.J.; Kim, H.W.; Klem, I.; Heitner, J.F.; Shah, D.J.; Jue, J.; White, B.E.; Indorkar, R.; et al. Feature-Tracking Global Longitudinal Strain Predicts Death in a Multicenter Population of Patients With Ischemic and Nonischemic Dilated Cardiomyopathy Incremental to Ejection Fraction and Late Gadolinium Enhancement. JACC Cardiovasc. Imaging 2018, 11, 1419–1429.
- Fatkin, D.; Huttner, I.G.; Kovacic, J.C.; Seidman, J.G.; Seidman, C.E. Precision Medicine in the Management of Dilated Cardiomyopathy: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2019, 74, 2921–2938.
- Haas, J.; Frese, K.S.; Peil, B.; Kloos, W.; Keller, A.; Nietsch, R.; Feng, Z.; Müller, S.; Kayvanpour, E.; Vogel, B.; et al. Atlas of the clinical genetics of human dilated cardiomyopathy. Eur. Heart J. 2015, 36, 1123–1135a.
- Merlo, M.; Stolfo, D.; Anzini, M.; Negri, F.; Pinamonti, B.; Barbati, G.; Ramani, F.; Lenarda, A.D.; Sinagra, G. Persistent recovery of normal left ventricular function and dimension in idiopathic dilated cardiomyopathy during long-term follow-up: Does real healing exist? J. Am. Heart Assoc. 2015, 4, e001504.
- Zakrzewska-Koperska, J.; Franaszczyk, M.; Bilińska, Z.; Truszkowska, G.; Karczmarz, M.; Szumowski, Ł.; Zieliński, T.; Płoski, R.; Bilińska, M. Rapid and effective response of the R222Q SCN5A to quinidine treatment in a patient with Purkinje-related ventricular arrhythmia and familial dilated cardiomyopathy: A case report. BMC Med. Genet. 2018, 19, 94.
- Rubiś, P.; Wiśniowska-Śmialek, S.; Wypasek, E.; Biernacka-Fijalkowska, B.; Rudnicka-Sosin, L.; Dziewiecka, E.; Faltyn, P.; Khachatryan, L.; Karabinowska, A.; Kozanecki, A.; et al. Fibrosis of extracellular matrix is related to the duration of the disease but is unrelated to the dynamics of collagen metabolism in dilated cardiomyopathy. Inflamm. Res. 2016, 65, 941–949.
- Kimura, Y.; Okumura, T.; Morimoto, R.; Kazama, S.; Shibata, N.; Oishi, H.; Araki, T.; Mizutani, T.; Kuwayama, T.; Hiraiwa, H.; et al. A clinical score for predicting left ventricular reverse remodelling in patients with dilated cardiomyopathy. ESC Heart Fail. 2021, 8, 1359–1368.
- Cannatà, A.; De Angelis, G.; Boscutti, A.; Normand, C.; Artico, J.; Gentile, P.; Zecchin, M.; Heymans, S.; Merlo, M.; Sinagra, G. Arrhythmic risk stratification in non-ischaemic dilated cardiomyopathy beyond ejection fraction. Heart 2020, 106, 656–664.
- Neilan, T.G.; Coelho-Filho, O.R.; Danik, S.B.; Shah, R.V.; Dodson, J.A.; Verdini, D.J.; Tokuda, M.; Daly, C.A.; Tedrow, U.B.; Stevenson, W.G.; et al. CMR quantification of myocardial scar provides additive prognostic information in nonischemic cardiomyopathy. JACC Cardiovasc. Imaging 2013, 6, 944–954.
- Barison, A.; Aimo, A.; Ortalda, A.; Todiere, G.; Grigoratos, C.; Passino, C.; Camici, P.G.; Aquaro, G.D.; Emdin, M. Late gadolinium enhancement as a predictor of functional recovery, need for defibrillator implantation and prognosis in non-ischemic dilated cardiomyopathy. Int. J. Cardiol. 2018, 250, 195–200.
- Merlo, M.; Masè, M.; Vitrella, G.; Belgrano, M.; Faganello, G.; Di Giusto, F.; Boscutti, A.; Gobbo, M.; Gigli, M.; Altinier, A.; et al. Usefulness of Addition of Magnetic Resonance Imaging to Echocardiographic Imaging to Predict Left Ventricular Reverse Remodeling in Patients With Nonischemic Cardiomyopathy. Am. J. Cardiol. 2018, 122, 490–497.
- Höke, U.; Khidir, M.J.; van der Geest, R.J.; Schalij, M.J.; Bax, J.J.; Delgado, V.; Ajmone Marsan, N. Relation of Myocardial Contrast-Enhanced T(1) Mapping by Cardiac Magnetic Resonance to Left Ventricular Reverse Remodeling After Cardiac Resynchronization Therapy in Patients With Nonischemic Cardiomyopathy. Am. J. Cardiol. 2017, 119, 1456–1462.
- Tobita, T.; Nomura, S.; Fujita, T.; Morita, H.; Asano, Y.; Onoue, K.; Ito, M.; Imai, Y.; Suzuki, A.; Ko, T.; et al. Genetic basis of cardiomyopathy and the genotypes involved in prognosis and left ventricular reverse remodeling. Sci. Rep. 2018, 8, 1998.