The overview of the origin of CNS macrophages has radically evolved during the last decades. In the past, all tissue-specific macrophages were considered to be derived from bone marrow progenitors [
17,
18,
19]. However, according to recent data from studies that have utilized new genetic tools, the early perception that tissue-resident macrophages derive solely from adult blood circulating monocytes is no longer prevalent [
8,
11,
20,
21,
22,
23,
24]. Although several studies have investigated the role of microglia in neuroinflammation and neurodegenerative diseases, such as Alzheimer’s disease, Parkinson’s disease and multiple sclerosis, little is known about the contribution of BAMs in these pathophysiological patterns [
25,
26,
27,
28,
29,
30]. There are apparent limitations to elucidating the contribution of BAMs in neurological disease; the exact pathophysiological role of BAMs and other CNS-innate immune cells cannot be interpreted by the majority of available tools; therefore, the distinction of BAMs from microglia under disease remains challenging [
12,
31]. Nevertheless, data obtained via contemporary technologies indicate their potential involvement and miscellaneous phenotypes in neurodegenerative and neuroinflammatory CNS diseases [
5,
12,
32,
33,
34,
35,
36].
2. Origin of BAMs during Embryogenesis and Adulthood
The BAM embryonic origin was first investigated in rodents using bone marrow chimeras and whole-body irradiation, proposing that BAMs are bone marrow-derived [
37,
38]. In 2016, Goldmann et al., performing fate mapping analysis, observed that BAMs originate from the mouse yolk sac’s early erythro-myeloid progenitors (EMPs) during embryogenesis [
8]. A tamoxifen-inducible Runx1
CreERR26
YFPfate-mapping mouse model confirmed that BAMs originate from early EMPs in the yolk sac, which gave rise to two different macrophage populations, namely, CD206
+ (BAM progenitors) and CD206
− (microglial progenitors) without the contribution of fetal liver or definitive hematopoiesis [
20]. Interestingly, the mannose receptor C-type 1 (MRC1 or CD206) is a unique marker for BAMs [
8,
12,
14]. The expression of
Mrc1 is upregulated from E8.5 when the primitive macrophages, originating from EMPs, prepare to invade the embryonic tissues [
39]. Recently, Masuda et al. investigated the progenitors of BAMs utilizing single-cell RNA sequencing and fate mapping analysis in the
Mrc1CreERT2 mouse model. Although flow cytometry confirmed the presence of a CD206
+ subpopulation within the A2 cells (CD45
+ c-kit
− CX
3CR1
+ cells), meningeal macrophages and microglia were found to originate from common CD206
+ A2 progenitors in contrast with previous results [
20,
40]. The pvΜΦ were generated postnatally from sdΜΦ, requiring integrin-signaling and vascular smooth muscle cells (VSMCs) [
40].
Regarding the repopulation pattern of BAMs in adulthood, there is a great heterogeneity between BAM clusters; specifically, the sdΜΦ, pvMΦ and cp
epiΜΦ exhibit similar longevity with microglial cells as being self-maintained in the CNS independently from blood monocytes’ contribution [
8,
14]. The cp
epiΜΦ were solely derived from local SALL1
+ macrophages [
14]. In
Ccr2-deficient mice, the number of cpMΦ decreased, revealing their replenishment from Ly6Chi monocytes and shorter turnover [
8]. In accordance with these results, Van Hove et al., combining single-cell RNA sequencing with complementary approaches in mice, suggested that dmΜΦ and cpΜΦ were gradually replenished by bone marrow-derived monocytes [
14]. As dura mater and choroid plexus stroma are more accessible brain regions than (i) subdural space, (ii) the apical surface of the choroid plexuses, and (iii) brain parenchyma, the tissue permeability may be considered a crucial factor for brain macrophage ontogeny. However, the ablation of BAMs through CSF1R blockade led to the replenishment of cpΜΦ and dmΜΦ via local expansion, indicating their self-renewal capacity, while sdΜΦ presented difficulties in their repopulation [
14].
By utilizing the Cx3cr1
CreER:R26
tdTomato fate mapping system in an experimental autoimmune encephalomyelitis (EAE) mouse model, Jordão et al. proposed that BAMs remained stable and locally self-renewed in addition to the recruitment of bone marrow-derived progenitors [
12]. In Cx3cr1
gfpCcr2
rfp bone marrow chimeric mice, CD169
+ BAMs proliferated after ischemia, while a small proportion of BAMs was bone marrow-derived, populating the perivascular and ischemic regions [
36]. Both in homeostasis and disease, skull and vertebrae bone marrow constitute a pool of myeloid cells that can invade non-parenchymal and parenchymal CNS regions, transforming into tissue-resident macrophages [
41]. A fate-mapping analysis in a mouse model of Alzheimer’s disease (AD) revealed that BAMs are a stable cell population with an unaffected turnover rate and a minimal replenishment from bone marrow-derived cells during this neurodegenerative disease [
42].
Summarizing, the origin of BAMs has been extensively studied in the last few years using new genetic tools, e.g., fate mapping analysis. It has been proposed that BAMs originate from early EMPs in the yolk sac during embryogenesis. Although specific BAMs are replenished by peripherally-derived monocytes postnatally, some remain solely derived from the local pool. BAMs have been shown to remain stable and locally self-renewed in both homeostasis and disease. Further investigation is needed to (i) confirm BAM origin, (ii) detect the precise embryonic progenitors of BAMs, especially of the dura mater and choroid plexus macrophages, (iii) determine the timing of each BAM subpopulation’s generation, and (iv) delineate their repopulation pattern.
3. Molecular Drives Orchestrating BAM Development
The transcription factor PU.1 (or SFPI) could be essential for the BAM generation during embryonic development since research has showed that in mice with deletion of the
Sfpi1 gene, pvMΦ, sdMΦ, and cpMΦ were ablated [
8]. Progenitors of BAMs express the runt-related transcription factor 1 (RUNX1), which regulates the expression of PU.1 during embryogenesis [
20,
43]. The impairment of PU.1 factor in mice results in a reduced number of A1 (CD45
+ c-kit
lo CX
3CR1
− immature cells) and A2 (CD45
+ c-kit
− CX
3CR1
+ cells) progenitor cells of the yolk sac, from which both microglial cells and BAMs originate. In contrast, the lack of interferon regulatory factor 8 (IRF8) exclusively decreased the number of A2 cells [
44]. Furthermore, the colony-stimulating factor 1 receptor (CSF1R) signaling could be essential for BAM development [
5,
11,
14]. In a zebrafish model carrying the panther mutation, a loss-of-function mutation in the
fms gene orthologue which encodes CSF1R, primitive macrophages of the yolk sac could not colonize the embryonic tissues [
45].
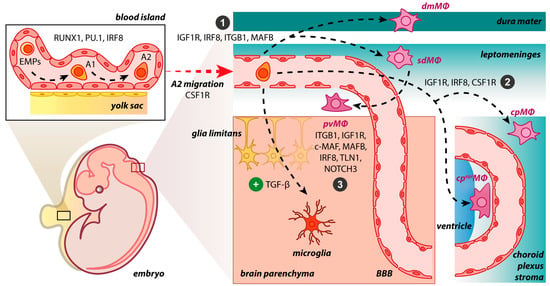
After progenitors’ migration and invasion in the CNS, BAM generation is initiated (
Figure 1). The BAMs may be developed independently of transforming growth factor beta receptor (TGF-βR) signaling. In
Tgfbr2-deficient mice, no alteration in cell numbers of BAMs occurred, while transforming growth factor beta (TGF-β) is required for the generation of microglial cells [
20,
46]. Three main brain border regions are filled with BAMs, namely, meninges, choroid plexus, and perivascular spaces. The postnatal expansion of sdMΦ was influenced by IRF8 and MAFB [
40]. Indeed, in
Irf8-deficient mice, a reduction of sdMΦ was observed [
8]. The lack of integrin subunit beta 1 (ITGB1) in mice resulted only in a minor change in the numbers of sdΜΦ [
40]. Similarly, the absence of insulin-like growth factor 1 (IGF1R) induces transcriptomic changes in BAMs via its implication in RNA processing, growth, migration and intracellular signaling [
47]. The MYB, BATF3, and NR4A1 transcription factors were not necessary for BAM development [
8].
Figure 1. BAM origin and propagation in the developing mouse brain. The early differentiation of macrophage progenitors is regulated by the expression of RUNX1, PU.1, and IRF8 in the yolk sac, where the primitive erythro-myeloid progenitors (EMPs) give rise to CD45+ c-kitlo CX3CR1− immature (A1) cells and subsequently to CD45+ c-kit− CX3CR1+ (A2) cells. In the presence of TGF-β, A2 cells initiate a microgliogenesis program upon settlement in the brain parenchyma. In the absence of the TGF-β, A2 cells do not enter the brain parenchyma, populate the abutting connective tissue, and may follow distinct developmental pathways: (1) IGF1R, IRF8, ITGB1, and MAFB restrict progenitors to the meninges, either dura or the subdural mesenchymal niche; (2) IGF1R, IRF8, and CSF1R dictate progenitors’ residency within the choroid plexus; (3) ITGB1, IGF1R, c-MAF, MAFB, IRF8, TLN1, and NOTCH3 stimulate pvΜΦ generation postnatally from sdΜΦ. However, it is not yet fully understood if the dmΜΦ share common progenitors and drivers with sdΜΦ during embryogenesis. dmΜΦ: dural macrophages; sdΜΦ: subdural macrophages; pvΜΦ: perivascular macrophages; cpΜΦ: stromal choroid plexus macrophages; cpepiΜΦ: choroid epiplexus macrophages; BBB: blood–brain barrier.
Specific molecular cues may also regulate the development of choroid plexus macrophages. Colony stimulating factor 1 (CSF1 or M-CSF), produced by stromal and epithelial cells, is crucial for macrophages’ ontogeny, orchestrating their proliferation and differentiation [
48]. CSF1 binds to its receptor, namely, CSF1R, a homodimeric type III receptor tyrosine kinase [
49]. Fms-intronic regulatory element (FIRE) is a highly conserved enhancer found in the second intron of the
Csf1r gene [
50]. In mutant mice with deletion of FIRE, the production and maintenance of cpMΦ were partially impaired [
51]. On the contrary, Rojo et al. demonstrated that in FIRE-deficient mice, microglial cells were absent from the brain, whereas BAMs were retained [
52]. Interestingly, cpMΦ remained unaltered in a study with
Irf8-deficient mice [
8], while other research considered IRF8 as a regulator of cpMΦ maturation since the gene ablation suppressed the transcriptional programme of cpMΦ [
14,
53].
The transcription factor c-MAF, a member of Maf family transcription factors, could be crucial for regulating the pvMΦ transcriptional programme as the deletion of
c-Maf in macrophage lineages resulted in the ablation of pvMΦ in the mouse brain [
54]. The postnatal expansion of pvΜΦ was also influenced by IRF8 and MAFB [
40]. Moreover, VSMCs have a potential role in the distribution of the pvMΦ during development. In
Notch3-deficient
mice, VSMCs are reduced similarly to the pvMΦ, while the number of sdΜΦ was maintained [
40,
55]. The distribution of pvMΦ is also controlled by integrin signaling.
Talin 1 (
Tln1) is an integrin-related gene which encodes a cytoskeletal protein. In
Tln1−/− mice, a significantly lower number of pvMΦ was observed, while microglia and sdΜΦ were not affected in the developing brain, underscoring the impaired vascularization as the cause of pvMΦ reduction [
40]. Nevertheless, the absence of integrin subunit beta 1 (ITGB1) in mice resulted only in a minor change in the numbers of pvΜΦ [
40].
To recapitulate, the emergence of BAMs is considered a complex process tightly regulated by multiple molecular cues in a similar pattern to oligodendrogenesis and microgliogenesis [
24,
56]. Although some molecular drivers orchestrating BAM generation have been recently discovered, it remains a largely uncharted territory.
4. BAMs vs. Microglia
Although microglia and BAMs are immune-competent cells of the CNS with common progenitors, their different localization may contribute to variations in their biological roles. The microglial populations’ functions have been reviewed in detail [
65,
66,
67]. Concisely, microglial cells are involved in developmental processes, including cell positioning, survival, myelinogenesis, synaptic patterning, and axonal dynamics [
68]. In adult CNS, microglia, as the regulators of acute and chronic immune responses, are implicated in removing pathogens and noxious particles, scavenging cellular debris and synapses, protecting neural tissue, and mediating neurogenesis in CNS injury [
24,
66,
69].
The unique localization of BAMs between brain parenchyma and peripheral tissues pinpoint their pivotal role in the immune surveillance of pathological antigens [
16]. Their antigen-presenting capacity is attributed to MHC II molecules on some BAM surfaces [
12,
32,
70,
71]. Furthermore, the pvΜΦ and dmΜΦ mainly phagocytose intruding pathogens and any foreign molecule or substance that can be detected in the bloodstream and cerebrospinal fluid [
72,
72]. The pvΜΦ also appear to regulate the accessibility of brain parenchyma to circulating cells and molecules by increasing the contractility of regional vessels and capillaries or diminishing the BBB permeability [
73,
74,
75,
76]. Interestingly, the latest approaches demonstrate the involvement of BAMs in ensuring a well-balanced metabolic environment for neurons, especially in the course of systemic perturbations [
77,
78].
Data regarding morphology, motility, and molecular identity of microglia and BAMs are summarized in Table 1.
Table 1. Morphology, motility, and specific surface markers of microglia and BAMs.
| Cell Type |
Morphology |
Motility |
Cell-Specific Markers |
| Microglia |
Ramified in homeostasis;
Amoeboid in inflammation |
Cell bodies with limited-motility but highly
dynamic processes in homeostasis;
Highly phagocytic in inflammation |
SIGLEC-H+, P2RY12+, HEXB+, TMEM119+, ANXA3+, SALL1+ |
| pvΜΦ |
Slightly elongated cell bodies |
Non-motile cell bodies with extending and retracting projections through the blood vessel wall in homeostasis;
Dendritic-like processes in inflammation |
CD206+, CD38+, LYVE1+, CD36+, CD163+, CD169+ |
| dmΜΦ |
Elongated; Spindle-shaped cells;
Few thick membrane projections; Dendriform |
Limited motility and highly dynamic
protrusions in homeostasis;
Extending projections in inflammation |
| sdΜΦ |
Elongated; Amoeboid;
Spindle-shaped cells;
Few thick membrane projections |
Limited motility and highly dynamic
protrusions in homeostasis;
Extending projections in inflammation |
| cpΜΦ |
Star-like shape |
Unknown |
| cpepiΜΦ |
Round; Bipolar; Stellate |
Unknown |
5. BAMs in Neurological Diseases and Promising Therapies
The implication of BAMs in the pathogenesis of CNS diseases, especially in neurodegeneration and neuroinflammation, is a rapidly emerging field of research. Although the precise role of BAMs in diseases is not yet elucidated, recent studies have addressed their potential involvement in several pathological conditions such as Alzheimer’s disease, Parkinson’s disease, multiple sclerosis, and stroke. Further experimental studies are needed to delineate the exact pathophysiological mechanism through which methodical manipulation of BAMs can halt or even reverse the progression of the aforementioned debilitating CNS diseases.
5.1. BAMs in Alzheimer’s Disease
AD is a brain disorder constituting a common cause of dementia, characterized by permanent neurodegeneration in specific brain areas [
86]. However, the pathophysiology of the disease is not yet fully understood. The accumulation of amyloid beta (Aβ) protein in the brain has been implicated in AD. This protein forms sticky plaques that may disrupt the interaction between brain cells, leading to inflammation and neuronal death [
87]. Additionally, AD is characterized by the accumulation of the tau protein, which forms neurofibrillary tangles [
88]. Patients with Alzheimer’s disease (AD) could be affected by cerebral amyloid angiopathy (CAA), which involves the pathologic deposition of Aβ within the leptomeningeal and cortical blood vessels [
89]. The role of pvΜΦ in CAA progression has been investigated in a TgCRND8 mouse model of AD [
90]. Hawkes and McLaurin demonstrated that the stimulation of the pvΜΦ turnover decreased cerebral CAA load. Interestingly, the clearance of CAA load was not attributed to microglia or astrocytes. These findings indicate the importance of pvΜΦ in CAA progression, suggesting that their activation could be a useful therapeutic approach for removing vascular amyloid [
90].
In Tg2576 mice, the clodronate-mediated depletion of pvΜΦ reduced the production of reactive oxygen species, thereby reversing cerebrovascular dysfunction induced by Aβ. Experiments utilizing bone marrow chimeras revealed that pvΜΦ are the primary cell expressing CD36 and NOX2, which are molecular substrates for inducing cerebrovascular oxidative stress [
91]. The pvΜΦ play a significant role in upregulating secreted phosphoprotein 1 (SPP1), with perivascular fibroblasts contributing to a lesser extent. SPP1 assists microglia in engulfing synapses and increases the expression of phagocytic markers such as complement C1q A chain (C1QA), granulin precursor (GRN), and cathepsin B (CTSB) in the presence of Aβ oligomers. The deletion of
Spp1 in AD mouse models prevented synaptic loss [
34]. Finally, the minor replenishment of CD206
+ BAMs and their stable turnover in a mouse AD model should be highlighted, as potential manipulations of these cells could lead to modification of AD pathology [
42].
5.2. BAMs in Parkinson’s Disease
Parkinson’s disease (PD) is another common neurodegenerative disorder characterized by dopaminergic cell loss [
92]. The accumulation of a-synuclein (α-SYN) is a distinct trait of degenerating dopaminergic neurons [
93,
94]. According to Guo et al., exosomes derived from microglia and CNS macrophages facilitated the transmission of α-SYN, leading to its aggregation in neurons and contributing to the development of PD [
95]. Interestingly, BAMs may mediate the α-SYN related neuroinflammation by acting as antigen-presenting cells essential for initiating a CD4 T cell response [
96]. The immune cell infiltration, recruitment, and antigen presentation were found to be greatly dependent on BAMs, framing their involvement in the pathogenesis of PD [
96]. A JAK1/2 inhibitor, namely, AZD1480, has been considered a therapeutic option for PD by reducing α-SYN-related neuroinflammation via downregulation of the JAK/STAT pathway [
97].
5.3. BAMs in Multiple Sclerosis
Multiple sclerosis (MS) is a debilitating neurodegenerative disease with a rising global prevalence in recent years [
98]. MS features encompass neuroinflammation, demyelination, and axonal loss within the CNS [
99,
100]. Several mechanisms have been proposed to be implicated in the pathophysiology of MS [
101,
102,
103]. Nevertheless, the potential role of BAMs in MS has been only recently investigated [
47,
104]. BAMs, as a CNS macrophage population, could potentially be involved in the MS course through the CNS-targeted autoimmunity or neurodegeneration leading to a secondary autoimmune response [
105]. BAMs are presented with different phenotypes regarding their roles in each stage of the MS [
106].
Particularly, Locatelli et al. identified various markers of BAMs, utilizing immunofluorescent techniques in a MS mouse model, as the neuroinflammatory lesions shifted from expansion to gradual resolution [
107]. In EAE, the most widely used animal model for studying MS aspects [
108,
109], antigen presentation and T cell reactivation were found to be regulated by both meningeal macrophages and microglia, revealing the involvement of BAMs in the disease [
110,
111]. The pvΜΦ and sdΜΦ were found to be modestly increased in the EAE mouse model, with sdΜΦ population expanding during disease onset, suggesting their implication in the initial acute phase of EAE. On the contrary, the sdΜΦ population decreased during the chronic phase of the disease and pvΜΦ proliferation remained unaltered [
12].
The BAMs could also exert miscellaneous functions in MS via interleukin 9 (IL9) upregulation. Donninelli et al. found that MS patients had higher IL9 levels in the cerebrospinal fluid obtained from post-mortem samples. Through flow cytometry of snap-frozen tissue blocks from the same patients’ brains, higher expression of IL9 was also observed in macrophages [
112]. Additionally, the disease-mediated peroxisome injury in BAMs, leading to demyelination and axonal loss, may be prevented through treatment with 4-Phenylbutyrate, which serves as a potential therapeutic approach for halting inflammatory demyelination and the progression of MS [
113]. Lastly, foamy macrophages are formed in brain regions during MS; by targeting lipophagy, remyelination can be promoted as some BAM subtypes may be involved in the aforementioned process [
114,
115].
5.4. BAMs in Other CNS Diseases
The BAMs have also been implicated in other CNS diseases, such as stroke. The study of Pedragosa et al. highlighted the major role of BAMs in different pathophysiological changes related to ischemic stroke, including the recruitment of granulocytes, increased expression of vascular endothelial growth factor (VEGF), and increased permeability of pial and cortical blood vessels [
116]. The induction of ischemic stroke resulted in the proliferation and migration of CD163
+ BAMs adopting a pro-inflammatory phenotype in the ischemic
rat parenchyma. Although CD169
+ perivascular macrophages were also observed to proliferate in response to ischemic stroke, they were replaced by infiltrating bone marrow-derived cells in mice. These findings were confirmed in a human model in which CD163+ cells were also accumulated in the ischemic region [
36]. In the subarachnoid hemorrhage (SAH), sdΜΦ and pvΜΦ are involved in erythrocyte uptake affecting the outcome of hemorrhage. Specifically, their depletion led to the reduction of large arterioles’ inflammation and microthrombosis after SAH [
117]. Ultimately, an induction of anti-inflammatory microglial/macrophage responses and subsequent neuroprotection could be achieved through the peripheral administration of interleukin 13 in cases of ischemic stroke [
118].