The misfolding and subsequent abnormal accumulation and aggregation of α-Synuclein (αSyn) as insoluble fibrils in Lewy bodies and Lewy neurites is the pathological hallmark of Parkinson’s disease (PD) and several neurodegenerative disorders. A combination of environmental and genetic factors is linked to αSyn misfolding, among which neuroinflammation is recognized to play an important role. Indeed, a number of studies indicate that a Toll-like receptor (TLR)-mediated neuroinflammation might lead to a dopaminergic neural loss, suggesting that TLRs could participate in the pathogenesis of PD as promoters of immune/neuroinflammatory responses.
- Alpha-synucleinopathies
- Toll-like receptors
- non-motor symptoms
1. Introduction
The term α-synucleinopathies defines a group of neurodegenerative disorders associated with α-Synuclein (αSyn) proteotoxicity and formation of protein aggregates in neurons and non-neuronal cells including microglia, pericytes, astrocytes, and oligodendrocytes [1]. Clinically, α-synucleinopathies comprise Lewy body disease (LBD) and multiple system atrophy (MSA). αSyn is a small protein composed by 140 amino acids belonging to the synucleins family (α-synuclein, β-synuclein, γ-synuclein, and synoretin), whose members share high sequence identity and expression pattern [2]. Although implicated in numerous cellular processes, the exact function of αSyn is still unclear. Under physiological conditions, it is soluble and may be involved in the compartmentalization, storage, and recycling of neurotransmitters, while pathological conditions can induce abnormal fold and formation of β-sheets, generate toxic and aggregated oligomers, and/or polymerize to produce intracellular plaques responsible for clinical signs [3]. In addition, nitration or cleavage are both associated with a greater tendency to aggregate [4][5][6].
Protein-protein interactions at the cytosolic level can influence the physiological and biochemical functions of αSyn and consequently its tendency to aggregate. This suggests that it is possible to modify the aggregation of αSyn by selectively regulating the expression of its protein partners [7][8]. Many patients with synucleinopathies experience motor symptoms that represent a hallmark of the disease. Motor symptoms are the result of a reduction in dopamine levels in the striatum and appear gradually and worsen over time as the disease progresses [9]. The specific symptoms and their severity can vary depending on the type of synucleinopathy and the progression of the disease can vary widely between individuals. In PD, it is estimated that up to 80% of dopaminergic cells in the substantia nigra may be lost before the characteristic motor symptoms become apparent [10][11].
2. Protein Aggregation and Propagation in Synucleinopathies
The proteostasis network works to maintain the proper balance between protein synthesis, folding, and degradation. It includes the ubiquitin-proteasome system, autophagy, chaperons, and heat shock proteins. Together, these pathways are responsible for regulating the number and the quality of proteins in the cell as well as their clearance. Perturbations in proteostasis have been associated with the pathogenesis of synucleinopathies [12]. αSyn is an intrinsically disordered protein that does not have a single, well-defined three-dimensional structure, but exists in a dynamic equilibrium of multiple conformations. It has been demonstrated that in certain conditions, such as oxidative stress, nitrosative stress, and inflammation, αSyn can misfold into amyloid-like fibrils. Once misfolded, αSyn can form cell-to-cell aggregates which can spread throughout the brain and cause further damage [12][13]. This hypothesis has been further supported by other studies, which have found that αSyn expression levels can influence the progression of diseases such as PD [14], Alzheimer disease (AD) [15], and Huntington’s disease (HD) [16]. Although it is still debated whether αSyn aggregation is the main leading cause for the neuronal death, to date, oligomeric/aggregated species of αSyn contained in the LBs and LNs are considered the hallmarks of PD and other synucleinopathies [17][18][19]. It has been proposed that the progression of Lewy pathology is from axon terminals to neuronal soma. However, a “mitocentric view of PD” claims that mitochondrial dysfunctions can lead to nigrostriatal degeneration regardless of Lewy pathology [20]. Studies have suggested that misfolding may be caused by exposure to toxins, age-related alterations in the αSyn structure, and gene mutations, and may cause an increased propensity to the formations of αSyn aggregates and fibrils [21][22]. αSyn aggregation initiates by a short sequence of a seed or native, partially folded or unfolded oligomers, which adopt a non-native conformation and auto-assemble into higher-order oligomers. These oligomers can serve as precursors of fibril nucleus, highly dynamic species that recruit other monomers eliciting a rapid polymerization into amyloid fibrils, hierarchical polymorphic stable structures derived from protofibrils that are responsible for the development of several diseases. The current goal is to identify the exact intermediate structures and pathways involved in certain diseases and that are characteristic for each individual. Since amyloid fibril-associated diseases are orphan drug, building a molecular profile for a group of individuals with certain feature clinical signs could improve the diagnosis and be helpful for the designing of molecules able to potentially prevent the pathological escalation.
Cell-to-cell propagation of αSyn pathology is also a feature of PD and related synucleinopathies [23][24][25][26]. Studies have found that misfolded αSyn can spread between neuroanatomically-connected regions of the brain, from neuron to neighboring cells such as other neurons, and microglial and astroglial cells in a prion-like manner. Dissemination of αSyn produces toxic aggregates, triggering a neuroinflammatory status that contributes to symptoms worsening over time and ultimately to neuronal death [26][27]. The internalization of αSyn seeds can also take place via transmembrane, transsynaptic endocytosis, phagocytosis, receptor-mediated endocytosis, cell injury and leaking, and tunneling nanotubes, Syn can activate microglia and other immune cells and promote the production of pro-inflammatory cytokines, suggesting that immune activation and αSyn accumulation may feed back into a positive loop, thus exacerbating the neurodegenerative process in PD and other synucleinopathies [28].
3. Toll-Like Receptors (TLRs) in α-Synuclein Aggregation
The Toll-like receptors (TLRs) represent a family (at least 10 members TLR1-TLR10) of transmembrane proteins expressed by immune and non-immune cells, including microglia, neurons, astrocytes, and oligodendrocytes involved in the activation of the innate immune system [29]. They are located on the surface of cells and recognize pathogen-associated molecular patterns (PAMPs) derived from bacteria, viruses, fungi, and other pathogens or can also be found in the endosome and cytoplasm where they detect and respond to viral nucleic acids [30][31]. TLRs are also able to recognize a wide variety of damage- or danger-associated molecular patterns (DAMPs), also known as alarmins, including αSyn, released by damaged neuronal cells and injured tissues [32].
TLRs and αSyn are shown to be reciprocally influenced in a positive feedback loop; indeed, αSyn increases the expression of TLRs, including TLR1, TLR2, TLR3, and the adaptor Myd88 [33] and TLR2 and TLR4 are dysregulated in PD patients and animal models [34][35][36][37]. TLR dysregulation has been linked to the accumulation of misfolded α-syn and thereby widely implicated in the pathogenesis of the synucleinopathies [30][38]. However, there is still controversy in synucleinopathies regarding the advantageous or detrimental functions of TLRs, especially of TLR4. For example, the lack of TLR4 is associated with αSyn upregulation [39] and dopamine depletion. Moreover, Stefanova et al. [40] showed that the ablation of LR4 prevented αSyn phagocytosis and clearance. However, another study using TLR4 knockout mice revealed less neuroinflammation and neurodegeneration [41]. Although most studies have focused on TLR2 and TLR4, this could also be true for other TLRs, including TLR7 and TLR9 [42]. In fact, TLR7 activation can lead to the accumulation of αSyn in the brain and TLR9 might also be involved in the activation of microglia and the production of pro-inflammatory cytokines in synucleinopathies [43]. The αSyn proteostasis in the nervous central system is controlled by a selective autophagy pathway termed “synucleinphagy” that also requires the presence of TLRs. TLR-mediated activation of microglia could engulf αSyn into autophagosomes to be degraded; therefore, the disruption of synucleinphagy can cause the accumulation of misfolded αSyn and neuronal death. Both TLR2 and TLR4 can interact with αSyn, promoting its internalization into microglia [40][44][45]. Furthermore, targeting TLR2 has proven to be a good approach for inhibiting pathogenic cell-to-cell αSyntransmission and astroglial inflammatory responses, and for clearing toxic species via autophagic machinery. Once internalized, αSyn oligomers spread from neuron to glial cells, engaging and activating TLRs on nearby microglia surface and directly modulating its uptake and intracellular trafficking. Therefore, targeting TLRs and reactive microglia may be a promising therapeutic strategy also for preventing cell-to-cell transmission and slowing the progression of synucleinopathies. Figure 1 reports a schematic representation of the TLRs’ involvement in αSyn-mediated neurodegeneration.
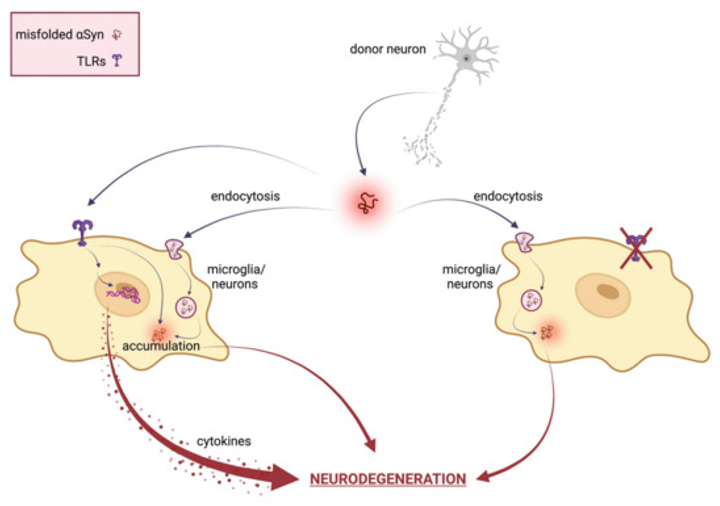
Figure 1. Schematical representation of TLRs involvement in αSyn mediated neurodegeneration. In neuronal cells, misfolded αSyn can be internalized via TLRs or several indirect endocytosis methods. Extracellular αSyn activates a TLR signaling cascade that results in neurotoxic responses, such as pro-inflammatory cytokine expression and release, ultimately leading to neuronal damage and pathological modification of αSyn. Therefore, TLR specific targeting ameliorates αSyn-mediated neurotoxicity by inhibiting TLR-mediated αSyn internalization and inflammatory response.
The characterization of the novel pathogenic species of αSyn and unexplored mechanisms that underlie their aggregation and spreading represent major goals and TLRs represent good candidates to look at. Furthermore, since the activation of specific TLR members on the cell surface or in intracellular milieu depend on specific αSyn conformations [46][47][48], associating a single TLR with a specific altered species of synuclein could help expand knowledge on the mechanisms of the synucleinopathies and help the designing of TLR agonists. In the plethora of animal models used to study PD and other neurodegenerative disorders, Drosophila melanogaster is taking the stage; indeed, the pathology observed in human PD can be accurately reproduced in Drosophila, with an area-specific and age-dependent loss of dopaminergic neurons, as well as LB and LN formation. Flies do not have the αSyn gene; however, the ectopic expression of human αSyn in the nervous system recapitulates cardinal features of Parkinson’s pathology [49], from the accumulation of protein aggregates to dopaminergic neurodegeneration, locomotor impairment, and lifespan reduction [50].
In Drosophila, the Toll family counts nine members (Toll, 18-Wheeler-, and Toll-3 to Toll-9), most of which are highly expressed during embryogenesis and metamorphosis and are involved in developmental regulation and in the onset of the immune response [51]. Drosophila TLRs do not function by recognizing different PAMP, as their mammalian counterparts do, and several of them exhibit also morphogenetic and neural functions [52]. The expression of Toll-related genes was analyzed in selected populations of cells across the brain: while Toll-1, Toll-2, Toll-6, Toll-7 and Toll-8exhibited a differential expression in the various neuronal subpopulations, Toll-4, Toll-5, and Toll-9 were not expressed by any of the CNS tested cells [53]. Evidence is increasing regarding the reciprocal relationship between Toll-mediated signaling pathway and neurodegeneration in Drosophila.
4. Non-Motor Symptoms in Synucleinopathies: A Focus on Circadian and Sleep Disturbances
The cardinal clinical feature of synucleinopathies, and neurodegenerative disorders in general, might be headed by a range of non-motor symptoms, such as cognitive impairment, autonomic dysfunction and circadian and sleep disturbances. These non-motor symptoms are increasingly relevant: on one hand they profoundly impact the patients’ quality of life and, on the other side, the fact that they precede motor symptoms and cognitive decline for many years suggests that in this pre-motor phase the pathogenic process is presumably underway and involves different regions of the peripheral and central nervous system [54]. Most human biological functions have specific temporal dynamics: biochemistry, physiology and behavior are temporally structured and characterized by a periodicity of about 24 h, in synchrony with the solar time. These rhythms are generated by an endogenous timekeeping system, the circadian clock, which regulates, among others, biological functions such as sleep/wake, feeding, body temperature, and hormonal levels. One of the prominent circadian-related symptoms in neurodegenerative disease patients is the alteration of sleep/wake patterns; during disease progression, nighttime sleep becomes progressively more fragmented, with a consequent increase in nocturnal activity and daytime sleepiness [55]. Molecular and physiological rhythms are also altered in PD patients, in which the normal 24-h oscillation in the clock gene Bmal1 expression was lost and melatonin levels were reduced [56]. Several studies have reported diurnal fluctuation in symptoms and signs associated with PD, such as motor symptoms [56], visual performance [57], and responsiveness to dopaminergic treatments [56], leading to the hypothesis of a circadian influence on the expression of clinical features of PD. The hypothesis of an intimate connection between circadian sleep regulation and α-synucleinopathies is further reinforced by the fact that melatonin was shown to prevent formation of αSyn aggregation as revealed by immunostaining in a cell model for maneb-induced neurodegeneration [58] and by a combination of several in vivo and in vitro approaches that revealed the ability of melatonin to affect the αSyn conformational dynamics and assembly, and consequently, its cytotoxicity [59].
5. Circadian and Sleep Disorders in α-Synucleinopathies: TLRs could Have a Say
Increasing evidence suggest that the circadian clock is closely associated with the immune system [60][61][62][63][64][65] and this association also involve TLRs [66]. Indeed, in vertebrates, the TLR9 gene expression is clock-controlled, with highest levels during the light phase and lower during the dark. This rhythmic expression is due to putative E-box sequences present in its promoter and ultimately results in a daily variation of the TLR9 mediated immune response [67]. In humans and animal models such as mouse and rat, the TLR4 ligand LPS can influence the expression of clock genes in the central pacemaker neurons, the suprachiasmatic nuclei, and peripherals; moreover, the levels of TLR4 were themselves reduced upon overexpression of the clock gene Cry1 [67][68]. TLR4 also mediates both the quality and the duration of sleep [68][69]; indeed, in light dark cycles, TLR4 KO mice exhibited an increased wakefulness in the early dark phase and a proportional reduction in the NREM sleep [68]. Both TLR2 and TLR4 contribute to sleep regulation: double TLR2/4 knock-out mice exhibit a 42% increase in REM sleep during daytime, and 41% more time awake during the night [69]. TLR2 is also involved in consolidating behavioral periodicity: young TLR2 KO mice show a reduced daily variability in rhythmic behaviors such movement, feeding, and drinking, in comparison to wild type [70]. A schematic representation of the reciprocal interaction involving TLRs, circadian machinery and neurodegeneration is reported in Figure 2.
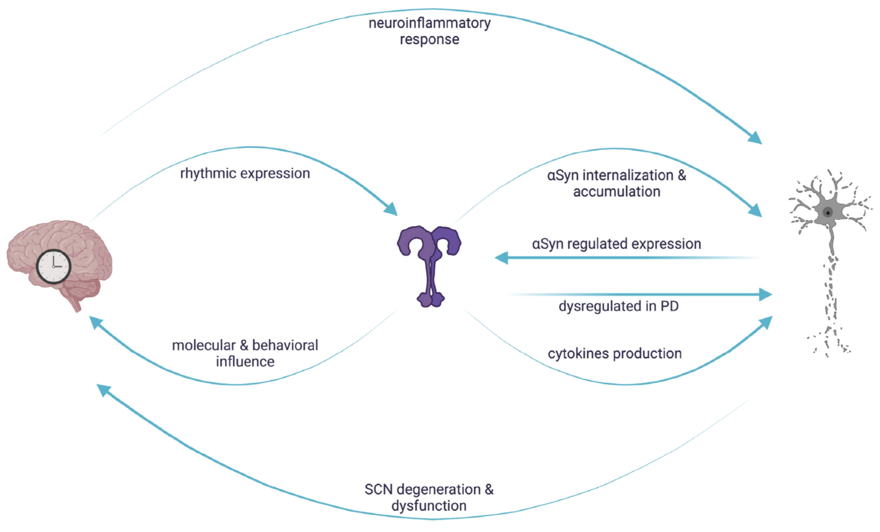
Figure 2. Reciprocal interaction between circadian regulation, TLR signaling and αSyn mediated neurodegeneration. In vertebrates, TLR expression is rhythmically controlled and in turn influences molecular and behavioral rhythms. Circadian clocks control the neuroinflammatory response leading to neuronal damage, which, in turn, causes circadian dysfunctions. The rhythmic regulation of TLRs could result in a daily variation of the downstream signaling cascades, conferring a circadian signature on αSyn-mediated neuronal degeneration.
Although loss of neuronal cells represents the primary cause of the development and progression of synucleinopathies, the underlying mechanisms are intricate. αSyn misfolding promotes the formation of insoluble structures that are believed to contribute to the disruption of cellular processes, neuronal death, and development of symptoms. Astrocytes, microglia, oligodendrocytes, and neurons orchestrate the complex crosstalk between intra- and extracellular αSyn levels by activation of intra- and extracellular TLRs. However, the finding regarding the beneficial or detrimental role of TLRs are conflicting and the precise molecular mechanisms are not completely understood. Further research is needed to clarify the relationship between TLRs and α-synucleinopathies and to determine whether TLR-targeted therapies may hold promise for treating these diseases. Moreover, as circadian and sleep disorders precede the emergence of cognitive and motor symptoms by years, the possibility to identify individuals at early stages of neurodegeneration will be an important step towards earlier intervention aimed at slowing the progression of the disease and minimizing its clinical disturbances.
This entry is adapted from the peer-reviewed paper 10.3390/cells12091231
References
- Stevenson, T.J.; Murray, H.C.; Turner, C.; Faull, R.L.M.; Dieriks, B. v.; Curtis, M.A. α-Synuclein Inclusions Are Abundant in Non-Neuronal Cells in the Anterior Olfactory Nucleus of the Parkinson’s Disease Olfactory Bulb. Sci Rep 2020, 10, 6682, doi:10.1038/s41598-020-63412-x.
- Clayton, D.F.; George, J.M. The Synucleins: A Family of Proteins Involved in Synaptic Function, Plasticity, Neurodegeneration and Disease. Trends Neurosci 1998, 21, 249–254, doi:10.1016/S0166-2236(97)01213-7.
- Soto, C.; Pritzkow, S. Protein Misfolding, Aggregation, and Conformational Strains in Neurodegenerative Diseases. Nat Neurosci 2018, 21, 1332–1340, doi:10.1038/s41593-018-0235-9.
- Zapadka, K.L.; Becher, F.J.; Gomes dos Santos, A.L.; Jackson, S.E. Factors Affecting the Physical Stability (Aggregation) of Peptide Therapeutics. Interface Focus 2017, 7, 20170030, doi:10.1098/rsfs.2017.0030.
- Burai, R.; Ait-Bouziad, N.; Chiki, A.; Lashuel, H.A. Elucidating the Role of Site-Specific Nitration of α-Synuclein in the Pathogenesis of Parkinson’s Disease via Protein Semisynthesis and Mutagenesis. J Am Chem Soc 2015, 137, 5041–5052, doi:10.1021/ja5131726.
- Liu, C.-W.; Giasson, B.I.; Lewis, K.A.; Lee, V.M.; DeMartino, G.N.; Thomas, P.J. A Precipitating Role for Truncated α-Synuclein and the Proteasome in α-Synuclein Aggregation. Journal of Biological Chemistry 2005, 280, 22670–22678, doi:10.1074/jbc.M501508200.
- Calabrese, G.; Molzahn, C.; Mayor, T. Protein Interaction Networks in Neurodegenerative Diseases: From Physiological Function to Aggregation. Journal of Biological Chemistry 2022, 298, 102062, doi:10.1016/j.jbc.2022.102062.
- Leitão, A.D.G.; Rudolffi-Soto, P.; Chappard, A.; Bhumkar, A.; Lau, D.; Hunter, D.J.B.; Gambin, Y.; Sierecki, E. Selectivity of Lewy Body Protein Interactions along the Aggregation Pathway of α-Synuclein. Commun Biol 2021, 4, 1124, doi:10.1038/s42003-021-02624-x.
- Franco, R.; Reyes-Resina, I.; Navarro, G. Dopamine in Health and Disease: Much More Than a Neurotransmitter. Biomedicines 2021, 9, 109, doi:10.3390/biomedicines9020109.
- Marsden, C.D. Parkinson’s Disease. The Lancet 1990, 335, 948–949, doi:10.1016/0140-6736(90)91006-V.
- Dauer, W.; Przedborski, S. Parkinson’s Disease. Neuron 2003, 39, 889–909, doi:10.1016/S0896-6273(03)00568-3.
- Lázaro, D.F.; Rodrigues, E.F.; Langohr, R.; Shahpasandzadeh, H.; Ribeiro, T.; Guerreiro, P.; Gerhardt, E.; Kröhnert, K.; Klucken, J.; Pereira, M.D.; et al. Systematic Comparison of the Effects of Alpha-Synuclein Mutations on Its Oligomerization and Aggregation. PLoS Genet 2014, 10, e1004741, doi:10.1371/journal.pgen.1004741.
- Cascella, R.; Bigi, A.; Cremades, N.; Cecchi, C. Effects of Oligomer Toxicity, Fibril Toxicity and Fibril Spreading in Synucleinopathies. Cellular and Molecular Life Sciences 2022, 79, 174, doi:10.1007/s00018-022-04166-9.
- Stefanis, L. -Synuclein in Parkinson’s Disease. Cold Spring Harb Perspect Med 2012, 2, a009399–a009399, doi:10.1101/cshperspect.a009399.
- Twohig, D.; Nielsen, H.M. α-Synuclein in the Pathophysiology of Alzheimer’s Disease. Mol Neurodegener 2019, 14, 23, doi:10.1186/s13024-019-0320-x.
- Pocas, G.M.; Branco-Santos, J.; Herrera, F.; Outeiro, T.F.; Domingos, P.M. -Synuclein Modifies Mutant Huntingtin Aggregation and Neurotoxicity in Drosophila. Hum Mol Genet 2015, 24, 1898–1907, doi:10.1093/hmg/ddu606.
- Wang, Z.; Becker, K.; Donadio, V.; Siedlak, S.; Yuan, J.; Rezaee, M.; Incensi, A.; Kuzkina, A.; Orrú, C.D.; Tatsuoka, C.; et al. Skin α-Synuclein Aggregation Seeding Activity as a Novel Biomarker for Parkinson Disease. JAMA Neurol 2021, 78, 30, doi:10.1001/jamaneurol.2020.3311.
- Shahnawaz, M.; Tokuda, T.; Waragai, M.; Mendez, N.; Ishii, R.; Trenkwalder, C.; Mollenhauer, B.; Soto, C. Development of a Biochemical Diagnosis of Parkinson Disease by Detection of α-Synuclein Misfolded Aggregates in Cerebrospinal Fluid. JAMA Neurol 2017, 74, 163, doi:10.1001/jamaneurol.2016.4547.
- El‐Agnaf, O.M.A.; Salem, S.A.; Paleologou, K.E.; Curran, M.D.; Gibson, M.J.; Court, J.A.; Schlossmacher, M.G.; Allsop, D. Detection of Oligomeric Forms of Α‐synuclein Protein in Human Plasma as a Potential Biomarker for Parkinson’s Disease. The FASEB Journal 2006, 20, 419–425, doi:10.1096/fj.03-1449com.
- Uchihara, T.; Giasson, B.I. Propagation of Alpha-Synuclein Pathology: Hypotheses, Discoveries, and yet Unresolved Questions from Experimental and Human Brain Studies. Acta Neuropathol 2016, 131, 49–73, doi:10.1007/s00401-015-1485-1.
- Ludtmann, M.H.R.; Angelova, P.R.; Horrocks, M.H.; Choi, M.L.; Rodrigues, M.; Baev, A.Y.; Berezhnov, A. V.; Yao, Z.; Little, D.; Banushi, B.; et al. α-Synuclein Oligomers Interact with ATP Synthase and Open the Permeability Transition Pore in Parkinson’s Disease. Nat Commun 2018, 9, 2293, doi:10.1038/s41467-018-04422-2.
- Trinh, J.; Zeldenrust, F.M.J.; Huang, J.; Kasten, M.; Schaake, S.; Petkovic, S.; Madoev, H.; Grünewald, A.; Almuammar, S.; König, I.R.; et al. Genotype-Phenotype Relations for the Parkinson’s Disease Genes SNCA, LRRK2, VPS35: MDSGene Systematic Review. Movement Disorders 2018, 33, 1857–1870, doi:10.1002/mds.27527.
- Ruiz-Riquelme, A.; Lau, H.H.C.; Stuart, E.; Goczi, A.N.; Wang, Z.; Schmitt-Ulms, G.; Watts, J.C. Prion-like Propagation of β-Amyloid Aggregates in the Absence of APP Overexpression. Acta Neuropathol Commun 2018, 6, 26, doi:10.1186/s40478-018-0529-x.
- Jan, A.; Gonçalves, N.P.; Vaegter, C.B.; Jensen, P.H.; Ferreira, N. The Prion-Like Spreading of Alpha-Synuclein in Parkinson’s Disease: Update on Models and Hypotheses. Int J Mol Sci 2021, 22, 8338, doi:10.3390/ijms22158338.
- Guo, J.L.; Lee, V.M.-Y. Seeding of Normal Tau by Pathological Tau Conformers Drives Pathogenesis of Alzheimer-like Tangles. Journal of Biological Chemistry 2011, 286, 15317–15331, doi:10.1074/jbc.M110.209296.
- Pearce, M.M.P.; Kopito, R.R. Prion-Like Characteristics of Polyglutamine-Containing Proteins. Cold Spring Harb Perspect Med 2018, 8, a024257, doi:10.1101/cshperspect.a024257.
- Sorrentino, Z.A.; Giasson, B.I.; Chakrabarty, P. α-Synuclein and Astrocytes: Tracing the Pathways from Homeostasis to Neurodegeneration in Lewy Body Disease. Acta Neuropathol 2019, 138, 1–21, doi:10.1007/s00401-019-01977-2.
- Kouli, A.; Horne, C.B.; Williams-Gray, C.H. Toll-like Receptors and Their Therapeutic Potential in Parkinson’s Disease and α-Synucleinopathies. Brain Behav Immun 2019, 81, 41–51, doi:10.1016/j.bbi.2019.06.042.
- Li, L.; Acioglu, C.; Heary, R.F.; Elkabes, S. Role of Astroglial Toll-like Receptors (TLRs) in Central Nervous System Infections, Injury and Neurodegenerative Diseases. Brain Behav Immun 2021, 91, 740–755, doi:10.1016/j.bbi.2020.10.007.
- Lind, N.A.; Rael, V.E.; Pestal, K.; Liu, B.; Barton, G.M. Regulation of the Nucleic Acid-Sensing Toll-like Receptors. Nat Rev Immunol 2022, 22, 224–235, doi:10.1038/s41577-021-00577-0.
- Conte, C. Possible Link between SARS-CoV-2 Infection and Parkinson’s Disease: The Role of Toll-Like Receptor 4. Int J Mol Sci 2021, 22, 7135, doi:10.3390/ijms22137135.
- Kawai, T.; Akira, S. Toll-like Receptors and Their Crosstalk with Other Innate Receptors in Infection and Immunity. Immunity 2011, 34, 637–650, doi:10.1016/j.immuni.2011.05.006.
- Béraud, D.; Twomey, M.; Bloom, B.; Mittereder, A.; Ton, V.; Neitzke, K.; Chasovskikh, S.; Mhyre, T.R.; Maguire-Zeiss, K.A. α-Synuclein Alters Toll-Like Receptor Expression. Front Neurosci 2011, 5, doi:10.3389/fnins.2011.00080.
- Gorecki, A.M.; Anyaegbu, C.C.; Anderton, R.S. TLR2 and TLR4 in Parkinson’s Disease Pathogenesis: The Environment Takes a Toll on the Gut. Transl Neurodegener 2021, 10, 47, doi:10.1186/s40035-021-00271-0.
- Dzamko, N.; Gysbers, A.; Perera, G.; Bahar, A.; Shankar, A.; Gao, J.; Fu, Y.; Halliday, G.M. Toll-like Receptor 2 Is Increased in Neurons in Parkinson’s Disease Brain and May Contribute to Alpha-Synuclein Pathology. Acta Neuropathol 2017, 133, 303–319, doi:10.1007/s00401-016-1648-8.
- Doorn, K.J.; Moors, T.; Drukarch, B.; van de Berg, W.D.; Lucassen, P.J.; van Dam, A.-M. Microglial Phenotypes and Toll-like Receptor 2 in the Substantia Nigra and Hippocampus of Incidental Lewy Body Disease Cases and Parkinson’s Disease Patients. Acta Neuropathol Commun 2014, 2, 90, doi:10.1186/s40478-014-0090-1.
- Zhang, W.; Wang, L.-Z.; Yu, J.-T.; Chi, Z.-F.; Tan, L. Increased Expressions of TLR2 and TLR4 on Peripheral Blood Mononuclear Cells from Patients with Alzheimer’s Disease. J Neurol Sci 2012, 315, 67–71, doi:10.1016/j.jns.2011.11.032.
- Kim, C.; Spencer, B.; Rockenstein, E.; Yamakado, H.; Mante, M.; Adame, A.; Fields, J.A.; Masliah, D.; Iba, M.; Lee, H.-J.; et al. Immunotherapy Targeting Toll-like Receptor 2 Alleviates Neurodegeneration in Models of Synucleinopathy by Modulating α-Synuclein Transmission and Neuroinflammation. Mol Neurodegener 2018, 13, 43, doi:10.1186/s13024-018-0276-2.
- Mariucci, G.; Pagiotti, R.; Galli, F.; Romani, L.; Conte, C. The Potential Role of Toll-Like Receptor 4 in Mediating Dopaminergic Cell Loss and Alpha-Synuclein Expression in the Acute MPTP Mouse Model of Parkinson’s Disease. Journal of Molecular Neuroscience 2018, 64, 611–618, doi:10.1007/s12031-018-1057-7.
- Stefanova, N.; Fellner, L.; Reindl, M.; Masliah, E.; Poewe, W.; Wenning, G.K. Toll-Like Receptor 4 Promotes α-Synuclein Clearance and Survival of Nigral Dopaminergic Neurons. Am J Pathol 2011, 179, 954–963, doi:10.1016/j.ajpath.2011.04.013.
- Perez-Pardo, P.; Dodiya, H.B.; Engen, P.A.; Forsyth, C.B.; Huschens, A.M.; Shaikh, M.; Voigt, R.M.; Naqib, A.; Green, S.J.; Kordower, J.H.; et al. Role of TLR4 in the Gut-Brain Axis in Parkinson’s Disease: A Translational Study from Men to Mice. Gut 2019, 68, 829–843, doi:10.1136/gutjnl-2018-316844.
- Heidari, A.; Yazdanpanah, N.; Rezaei, N. The Role of Toll-like Receptors and Neuroinflammation in Parkinson’s Disease. J Neuroinflammation 2022, 19, 135, doi:10.1186/s12974-022-02496-w.
- Fiebich, B.L.; Batista, C.R.A.; Saliba, S.W.; Yousif, N.M.; de Oliveira, A.C.P. Role of Microglia TLRs in Neurodegeneration. Front Cell Neurosci 2018, 12, doi:10.3389/fncel.2018.00329.
- Rannikko, E.H.; Weber, S.S.; Kahle, P.J. Exogenous α-Synuclein Induces Toll-like Receptor 4 Dependent Inflammatory Responses in Astrocytes. BMC Neurosci 2015, 16, 57, doi:10.1186/s12868-015-0192-0.
- Fellner, L.; Irschick, R.; Schanda, K.; Reindl, M.; Klimaschewski, L.; Poewe, W.; Wenning, G.K.; Stefanova, N. Toll‐like Receptor 4 Is Required for Α‐synuclein Dependent Activation of Microglia and Astroglia. Glia 2013, 61, 349–360, doi:10.1002/glia.22437.
- Codolo, G.; Plotegher, N.; Pozzobon, T.; Brucale, M.; Tessari, I.; Bubacco, L.; de Bernard, M. Triggering of Inflammasome by Aggregated α–Synuclein, an Inflammatory Response in Synucleinopathies. PLoS One 2013, 8, e55375, doi:10.1371/journal.pone.0055375.
- Daniele, S.G.; Béraud, D.; Davenport, C.; Cheng, K.; Yin, H.; Maguire-Zeiss, K.A. Activation of MyD88-Dependent TLR1/2 Signaling by Misfolded α-Synuclein, a Protein Linked to Neurodegenerative Disorders. Sci Signal 2015, 8, doi:10.1126/scisignal.2005965.
- Kim, C.; Ho, D.-H.; Suk, J.-E.; You, S.; Michael, S.; Kang, J.; Joong Lee, S.; Masliah, E.; Hwang, D.; Lee, H.-J.; et al. Neuron-Released Oligomeric α-Synuclein Is an Endogenous Agonist of TLR2 for Paracrine Activation of Microglia. Nat Commun 2013, 4, 1562, doi:10.1038/ncomms2534.
- Feany, M.B.; Bender, W.W. A Drosophila Model of Parkinson’s Disease. Nature 2000, 404, 394–398, doi:10.1038/35006074.
- Agostini, F.; Bubacco, L.; Chakrabarti, S.; Bisaglia, M. α-Synuclein Toxicity in Drosophila Melanogaster Is Enhanced by the Presence of Iron: Implications for Parkinson’s Disease. Antioxidants 2023, 12, 261, doi:10.3390/antiox12020261.
- Bilak, H.; Tauszig-Delamasure, S.; Imler, J.-L. Toll and Toll-like Receptors in Drosophila. Biochem Soc Trans 2003, 31, 648–651, doi:10.1042/bst0310648.
- Brennan, C.A.; Anderson, K. V. Drosophila: The Genetics of Innate Immune Recognition and Response. Annu Rev Immunol 2004, 22, 457–483, doi:10.1146/annurev.immunol.22.012703.104626.
- Zhan, L.; Xie, Q.; Tibbetts, R.S. Opposing Roles of P38 and JNK in a Drosophila Model of TDP-43 Proteinopathy Reveal Oxidative Stress and Innate Immunity as Pathogenic Components of Neurodegeneration. Hum Mol Genet 2015, 24, 757–772, doi:10.1093/hmg/ddu493.
- Raza, C.; Anjum, R.; Shakeel, N. ul A. Parkinson’s Disease: Mechanisms, Translational Models and Management Strategies. Life Sci 2019, 226, 77–90, doi:10.1016/j.lfs.2019.03.057.
- Hood, S.; Amir, S. The Aging Clock: Circadian Rhythms and Later Life. Journal of Clinical Investigation 2017, 127, 437–446, doi:10.1172/JCI90328.
- Bonuccelli, U.; Del Dotto, P.; Lucetti, C.; Petrozzi, L.; Bernardini, S.; Gambaccini, G.; Rossi, G.; Piccini, P. Diurnal Motor Variations to Repeated Doses of Levodopa in Parkinson’s Disease. Clin Neuropharmacol 2000, 23, 28–33, doi:10.1097/00002826-200001000-00006.
- Struck, L.K.; Rodnitzky, R.L.; Dobson, J.K. Stroke and Its Modification in Parkinson’s Disease. Stroke 1990, 21, 1395–1399, doi:10.1161/01.STR.21.10.1395.
- Ono, K.; Mochizuki, H.; Ikeda, T.; Nihira, T.; Takasaki, J.; Teplow, D.B.; Yamada, M. Effect of Melatonin on α-Synuclein Self-Assembly and Cytotoxicity. Neurobiol Aging 2012, 33, 2172–2185, doi:10.1016/j.neurobiolaging.2011.10.015.
- Lauretti, E.; Di Meco, A.; Merali, S.; Praticò, D. Circadian Rhythm Dysfunction: A Novel Environmental Risk Factor for Parkinson’s Disease. Mol Psychiatry 2017, 22, 280–286, doi:10.1038/mp.2016.47.
- Abele, S.H.; Meadows, K.E.; Medeiros, D.; Silver, A.C. Time Is on the Immune System’s Side, Yes It Is. Yale J Biol Med 2019, 92, 225–231.
- Labrecque, N.; Cermakian, N. Circadian Clocks in the Immune System. J Biol Rhythms 2015, 30, 277–290, doi:10.1177/0748730415577723.
- Hergenhan, S.; Holtkamp, S.; Scheiermann, C. Molecular Interactions Between Components of the Circadian Clock and the Immune System. J Mol Biol 2020, 432, 3700–3713, doi:10.1016/j.jmb.2019.12.044.
- Waggoner, S.N. Circadian Rhythms in Immunity. Curr Allergy Asthma Rep 2020, 20, 2, doi:10.1007/s11882-020-0896-9.
- Arjona, A.; Silver, A.C.; Walker, W.E.; Fikrig, E. Immunity’s Fourth Dimension: Approaching the Circadian–Immune Connection. Trends Immunol 2012, 33, 607–612, doi:10.1016/j.it.2012.08.007.
- Fan, X.; Song, Y.; Qin, D.-X.; Lin, P.-Y. Regulatory Effects of Clock and Bmal1 on Circadian Rhythmic TLR Expression. Int Rev Immunol 2023, 42, 101–112, doi:10.1080/08830185.2021.1931170.
- Silver, A.C.; Arjona, A.; Walker, W.E.; Fikrig, E. The Circadian Clock Controls Toll-like Receptor 9-Mediated Innate and Adaptive Immunity. Immunity 2012, 36, 251–261, doi:10.1016/j.immuni.2011.12.017.
- De Pablo-Fernández, E.; Courtney, R.; Warner, T.T.; Holton, J.L. A Histologic Study of the Circadian System in Parkinson Disease, Multiple System Atrophy, and Progressive Supranuclear Palsy. JAMA Neurol 2018, 75, 1008, doi:10.1001/jamaneurol.2018.0640.
- Wisor, J.P.; Clegern, W.C.; Schmidt, M.A. Toll-Like Receptor 4 Is a Regulator of Monocyte and Electroencephalographic Responses to Sleep Loss. Sleep 2011, 34, 1335–1345, doi:10.5665/SLEEP.1274.
- Sartorius, T.; Lutz, S.Z.; Hoene, M.; Waak, J.; Peter, A.; Weigert, C.; Rammensee, H.; Kahle, P.J.; Häring, H.; Hennige, A.M. Toll‐like Receptors 2 and 4 Impair Insulin‐mediated Brain Activity by Interleukin‐6 and Osteopontin and Alter Sleep Architecture. The FASEB Journal 2012, 26, 1799–1809, doi:10.1096/fj.11-191023.
- DeKorver, N.W.; Chaudoin, T.R.; Bonasera, S.J. Toll-Like Receptor 2 Is a Regulator of Circadian Active and Inactive State Consolidation in C57BL/6 Mice. Front Aging Neurosci 2017, 9, doi:10.3389/fnagi.2017.00219.