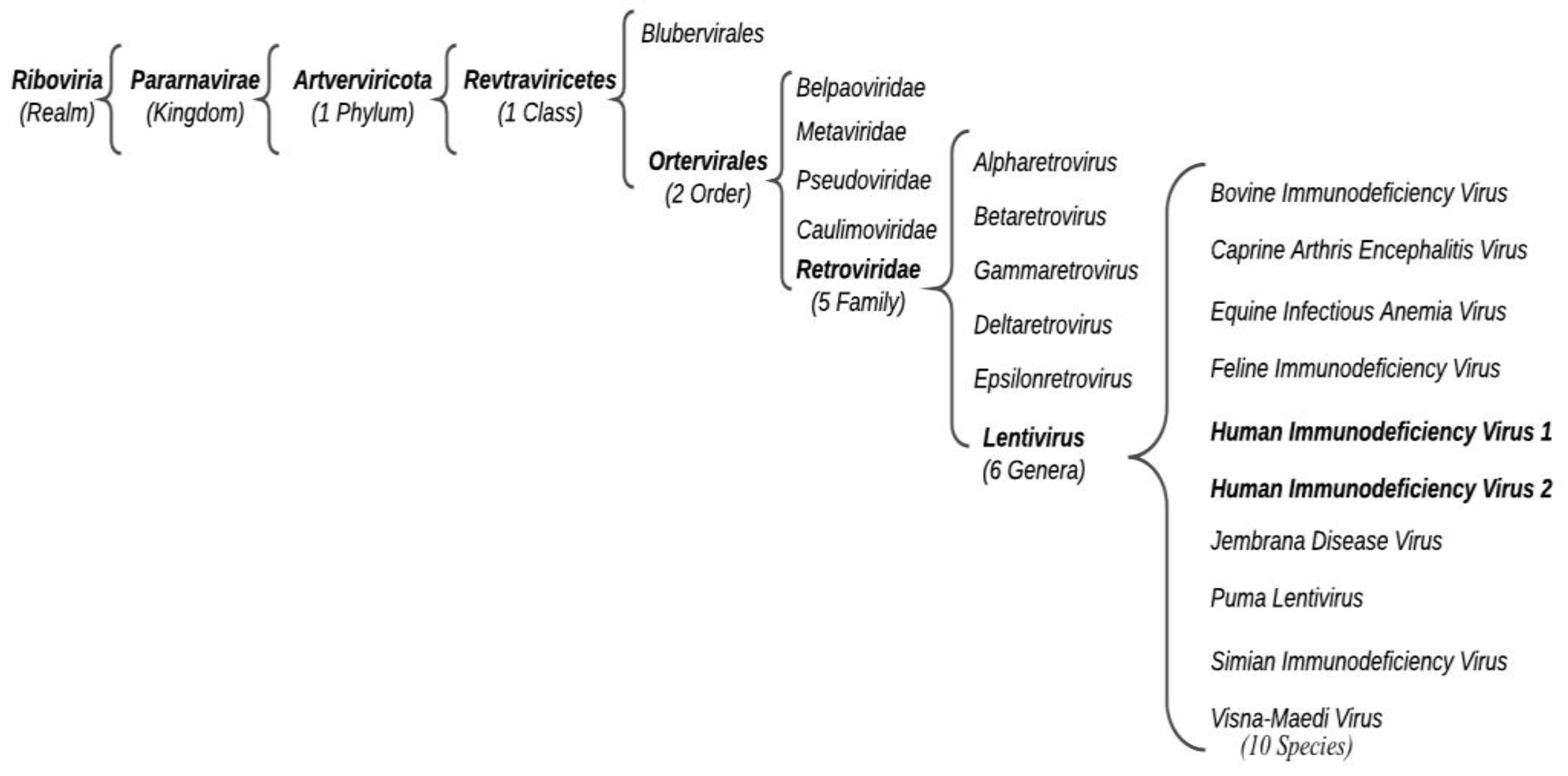
Acquired Immunodeficiency Syndrome (AIDS) is a human viral infectious disease caused by the positive-sense single-stranded (ss) RNA Human Immunodeficiency Virus (HIV) (Retroviridae family, Ortervirales order). HIV-1 can be distinguished into various worldwide spread groups and subtypes. HIV-2 also causes human immunodeficiency, which develops slowly and tends to be less aggressive. HIV-2 only partially homologates to HIV-1 despite the similar derivation. Antiretroviral therapy (ART) is the treatment approved to control HIV infection, based on multiple antiretroviral drugs that belong to different classes: (i) NNRTIs, (ii) NRTIs, (iii) PIs, (iv) INSTIs, and (v) entry inhibitors. These drugs, acting on different stages of the HIV life cycle, decrease the patient’s total burden of HIV, maintain the function of the immune system, and prevent opportunistic infections.
- HIV
- ART
- drug resistance
- retrovirus
- NNRTIs
- NRTIs
- PIs
- INSTIs
1. Introduction: HIV and Its Distribution
2. HIV Diagnosis and Clinical Course
3. Treatment
4. Drug Resistance Mechanism
5. NRTI Drug Resistance
6. NNRTIs Drugs Resistance
7. PIs Drugs Resistance
8. INSTIs Drugs Resistance
9. Entry Inhibitor Drugs Resistance
9.1. Chemokine Receptor 5 (CCR5) Antagonists
9.2. Entry Inhibitor of gp41
10. Conclusions
This entry is adapted from the peer-reviewed paper 10.3390/microorganisms11010221
References
- Bbosa, N.; Kaleebu, P.; Ssemwanga, D. HIV Subtype Diversity Worldwide. Curr. Opin. HIV AIDS 2019, 14, 153–160.
- Gallo, R.C.; Montagnier, L. The Discovery of HIV as the Cause of AIDS. N. Engl. J. Med. 2003, 349, 2283–2285.
- Montagnier, L. 25 Years after HIV Discovery: Prospects for Cure and Vaccine. Virology 2010, 397, 248–254.
- Vahlne, A. A Historical Reflection on the Discovery of Human Retroviruses. Retrovirology 2009, 6, 40.
- Hemelaar, J.; Gouws, E.; Ghys, P.D.; Osmanov, S. Global and Regional Distribution of HIV-1 Genetic Subtypes and Recombinants in 2004. AIDS 2006, 20, W13–W23.
- Sharp, P.M.; Hahn, B.H. Origins of HIV and the AIDS Pandemic. Cold Spring Harb. Perspect. Med. 2011, 1, a006841.
- Vidal, N.; Peeters, M.; Mulanga-Kabeya, C.; Nzilambi, N.; Robertson, D.; Ilunga, W.; Sema, H.; Tshimanga, K.; Bongo, B.; Delaporte, E. Unprecedented Degree of Human Immunodeficiency Virus Type 1 (HIV-1) Group M Genetic Diversity in the Democratic Republic of Congo Suggests That the HIV-1 Pandemic Originated in Central Africa. J. Virol. 2000, 74, 10498–10507.
- Casado, G.; Thomson, M.M.; Sierra, M.; Nájera, R. Identification of a Novel HIV-1 Circulating ADG Intersubtype Recombinant Form (CRF19_cpx) in Cuba. J. Acquir. Immune Defic. Syndr. 2005, 40, 532–537.
- Ng, K.T.; Ong, L.Y.; Takebe, Y.; Kamarulzaman, A.; Tee, K.K. Genome Sequence of a Novel HIV-1 Circulating Recombinant Form 54_01B from Malaysia. J. Virol. 2012, 86, 11405–11406.
- Song, H.; Giorgi, E.E.; Ganusov, V.V.; Cai, F.; Athreya, G.; Yoon, H.; Carja, O.; Hora, B.; Hraber, P.; Romero-Severson, E.; et al. Tracking HIV-1 Recombination to Resolve Its Contribution to HIV-1 Evolution in Natural Infection. Nat. Commun. 2018, 9, 1928.
- Osmanov, S.; Pattou, C.; Walker, N.; Schwardländer, B.; Esparza, J. WHO-UNAIDS Network for HIV Isolation and Characterization Estimated Global Distribution and Regional Spread of HIV-1 Genetic Subtypes in the Year 2000. J. Acquir. Immune Defic. Syndr. 2002, 29, 184–190.
- Hanna, G.J.; Balaguera, H.U.; Freedberg, K.A.; Werner, B.G.; Steger Craven, K.A.; Craven, D.E.; D’Aquila, R.T. Drug-Selected Resistance Mutations and Non-B Subtypes in Antiretroviral-Naive Adults with Established Human Immunodeficiency Virus Infection. J. Infect. Dis. 2003, 188, 986–991.
- Rhee, S.-Y.; Shafer, R.W. Geographically-Stratified HIV-1 Group M Pol Subtype and Circulating Recombinant Form Sequences. Sci. Data 2018, 5, 180148.
- Gartner, M.J.; Roche, M.; Churchill, M.J.; Gorry, P.R.; Flynn, J.K. Understanding the Mechanisms Driving the Spread of Subtype C HIV-1. EBioMedicine 2020, 53, 102682.
- Khan, S.; Zahid, M.; Qureshi, M.A.; Mughal, M.N.; Ujjan, I.D. HIV-1 Genetic Diversity, Geographical Linkages and Antiretroviral Drug Resistance among Individuals from Pakistan. Arch. Virol. 2018, 163, 33–40.
- Korber, B.; Gaschen, B.; Yusim, K.; Thakallapally, R.; Kesmir, C.; Detours, V. Evolutionary and Immunological Implications of Contemporary HIV-1 Variation. Br. Med. Bull. 2001, 58, 19–42.
- Menéndez-Arias, L.; Alvarez, M. Antiretroviral Therapy and Drug Resistance in Human Immunodeficiency Virus Type 2 Infection. Antivir. Res. 2014, 102, 70–86.
- Lu, H.; Tang, Y.-W. Myths in the Laboratory Diagnosis of HIV Infection. Emerg. Microbes Infect. 2019, 8, 1240–1242.
- Hurt, C.B.; Nelson, J.A.E.; Hightow-Weidman, L.B.; Miller, W.C. Selecting an HIV Test: A Narrative Review for Clinicians and Researchers. Sex. Transm. Dis. 2017, 44, 739–746.
- Alexander, T.S. Human Immunodeficiency Virus Diagnostic Testing: 30 Years of Evolution. Clin. Vaccine Immunol. 2016, 23, 249–253.
- Pitasi, M.A.; Patel, S.N.; Wesolowski, L.G.; Masciotra, S.; Luo, W.; Owen, S.M.; Delaney, K.P. Performance of an Alternative Laboratory-Based HIV Diagnostic Testing Algorithm Using HIV-1 RNA Viral Load. Sex. Transm. Dis. 2020, 47, S18–S25.
- Robertson, M.M.; Braunstein, S.L.; Hoover, D.R.; Li, S.; Nash, D. Timeliness of Human Immunodeficiency Virus Diagnosis and Antiretroviral Treatment Initiation in the Era of Universal Testing and Treatment. J. Infect. Dis. 2019, 220, 648–656.
- Yang, O.O.; Cumberland, W.G.; Escobar, R.; Liao, D.; Chew, K.W. Demographics and Natural History of HIV-1-Infected Spontaneous Controllers of Viremia. AIDS 2017, 31, 1091–1098.
- Platten, M.; Linnemann, R.; Kümmerle, T.; Jung, N.; Wyen, C.; Ehren, K.; Gravemann, S.; Gillor, D.; Cornely, O.A.; Fischer, J.; et al. Clinical Course and Quality of Care in ART-Naïve Patients Newly Presenting in a HIV Outpatient Clinic. Infection 2014, 42, 849–857.
- Sneller, M.C.; Blazkova, J.; Justement, J.S.; Shi, V.; Kennedy, B.D.; Gittens, K.; Tolstenko, J.; McCormack, G.; Whitehead, E.J.; Schneck, R.F.; et al. Combination Anti-HIV Antibodies Provide Sustained Virological Suppression. Nature 2022, 606, 375–381.
- Merigan, T.C. Treatment of AIDS with Combinations of Antiretroviral Agents. Am. J. Med. 1991, 90, 8S–17S.
- Nastri, B.M.; Zannella, C.; Folliero, V.; Rinaldi, L.; Restivo, L.; Stelitano, D.; Sperlongano, R.; Adinolfi, L.E.; Franci, G. Editorial-Role of Highly Active Antiretroviral Therapy (HAART) for the COVID-19 Treatment. Eur. Rev. Med. Pharm. Sci. 2020, 24, 11982–11984.
- Sanna, G.; Madeddu, S.; Murgia, G.; Serreli, G.; Begala, M.; Caboni, P.; Incani, A.; Franci, G.; Galdiero, M.; Giliberti, G. Potent and Selective Activity against Human Immunodeficiency Virus 1 (HIV-1) of Thymelaea Hirsuta Extracts. Viruses 2020, 12, E664.
- Scott, L.J. Dolutegravir/Lamivudine Single-Tablet Regimen: A Review in HIV-1 Infection. Drugs 2020, 80, 61–72.
- Sarafianos, S.G.; Hughes, S.H.; Arnold, E. Designing Anti-AIDS Drugs Targeting the Major Mechanism of HIV-1 RT Resistance to Nucleoside Analog Drugs. Int. J. Biochem. Cell Biol. 2004, 36, 1706–1715.
- Voshavar, C. Protease Inhibitors for the Treatment of HIV/AIDS: Recent Advances and Future Challenges. Curr. Top. Med. Chem. 2019, 19, 1571–1598.
- Farady, C.J.; Craik, C.S. Mechanisms of Macromolecular Protease Inhibitors. ChemBiochem 2010, 11, 2341–2346.
- Ghosh, A.K.; Osswald, H.L.; Prato, G. Recent Progress in the Development of HIV-1 Protease Inhibitors for the Treatment of HIV/AIDS. J. Med. Chem. 2016, 59, 5172–5208.
- Brik, A.; Wong, C.-H. HIV-1 Protease: Mechanism and Drug Discovery. Org. Biomol. Chem. 2003, 1, 5–14.
- Anstett, K.; Brenner, B.; Mesplede, T.; Wainberg, M.A. HIV Drug Resistance against Strand Transfer Integrase Inhibitors. Retrovirology 2017, 14, 36.
- Delelis, O.; Carayon, K.; Saïb, A.; Deprez, E.; Mouscadet, J.-F. Integrase and Integration: Biochemical Activities of HIV-1 Integrase. Retrovirology 2008, 5, 114.
- Quashie, P.K.; Mesplède, T.; Wainberg, M.A. HIV Drug Resistance and the Advent of Integrase Inhibitors. Curr. Infect. Dis. Rep. 2013, 15, 85–100.
- Esté, J.A.; Telenti, A. HIV Entry Inhibitors. Lancet 2007, 370, 81–88.
- Pugach, P.; Ketas, T.J.; Michael, E.; Moore, J.P. Neutralizing Antibody and Anti-Retroviral Drug Sensitivities of HIV-1 Isolates Resistant to Small Molecule CCR5 Inhibitors. Virology 2008, 377, 401–407.
- Qian, K.; Morris-Natschke, S.L.; Lee, K.-H. HIV Entry Inhibitors and Their Potential in HIV Therapy. Med. Res. Rev. 2009, 29, 369–393.
- Dvory-Sobol, H.; Shaik, N.; Callebaut, C.; Rhee, M.S. Lenacapavir: A First-in-Class HIV-1 Capsid Inhibitor. Curr. Opin. HIV AIDS 2022, 17, 15–21.
- Diamond, T.L.; Ngo, W.; Xu, M.; Goh, S.L.; Rodriguez, S.; Lai, M.-T.; Asante-Appiah, E.; Grobler, J.A. Islatravir Has a High Barrier to Resistance and Exhibits a Differentiated Resistance Profile from Approved Nucleoside Reverse Transcriptase Inhibitors (NRTIs). Antimicrob. Agents Chemother. 2022, 66, e00133-22.
- Cane, P.A. Stability of Transmitted Drug-Resistant HIV-1 Species. Curr. Opin. Infect. Dis. 2005, 18, 537–542.
- Kitayimbwa, J.M.; Mugisha, J.Y.T.; Saenz, R.A. Estimation of the HIV-1 Backward Mutation Rate from Transmitted Drug-Resistant Strains. Popul. Biol. 2016, 112, 33–42.
- Abram, M.E.; Ferris, A.L.; Shao, W.; Alvord, W.G.; Hughes, S.H. Nature, Position, and Frequency of Mutations Made in a Single Cycle of HIV-1 Replication. J. Virol. 2010, 84, 9864–9878.
- Mansky, L.M. HIV Mutagenesis and the Evolution of Antiretroviral Drug Resistance. Drug Resist. Updat. 2002, 5, 219–223.
- Keele, B.F.; Giorgi, E.E.; Salazar-Gonzalez, J.F.; Decker, J.M.; Pham, K.T.; Salazar, M.G.; Sun, C.; Grayson, T.; Wang, S.; Li, H.; et al. Identification and Characterization of Transmitted and Early Founder Virus Envelopes in Primary HIV-1 Infection. Proc. Natl. Acad. Sci. USA 2008, 105, 7552–7557.
- Nasir, A.; Dimitrijevic, M.; Romero-Severson, E.; Leitner, T. Large Evolutionary Rate Heterogeneity among and within HIV-1 Subtypes and CRFs. Viruses 2021, 13, 1689.
- Levy, D.N.; Aldrovandi, G.M.; Kutsch, O.; Shaw, G.M. Dynamics of HIV-1 Recombination in Its Natural Target Cells. Proc. Natl. Acad. Sci. USA 2004, 101, 4204–4209.
- Hammer, S.M.; Saag, M.S.; Schechter, M.; Montaner, J.S.G.; Schooley, R.T.; Jacobsen, D.M.; Thompson, M.A.; Carpenter, C.C.J.; Fischl, M.A.; Gazzard, B.G.; et al. Treatment for Adult HIV Infection: 2006 Recommendations of the International AIDS Society-USA Panel. JAMA 2006, 296, 827–843.
- Coffin, J.M. HIV Population Dynamics in Vivo: Implications for Genetic Variation, Pathogenesis, and Therapy. Science 1995, 267, 483–489.
- Miller, R.L.; Ponte, R.; Jones, B.R.; Kinloch, N.N.; Omondi, F.H.; Jenabian, M.-A.; Dupuy, F.P.; Fromentin, R.; Brassard, P.; Mehraj, V.; et al. HIV Diversity and Genetic Compartmentalization in Blood and Testes during Suppressive Antiretroviral Therapy. J. Virol. 2019, 93, e00755-19.
- Perelson, A.S.; Ribeiro, R.M. Modeling the Within-Host Dynamics of HIV Infection. BMC Biol. 2013, 11, 96.
- Tang, M.W.; Shafer, R.W. HIV-1 Antiretroviral Resistance: Scientific Principles and Clinical Applications. Drugs 2012, 72, e1–e25.
- Sun, Z.; Lan, Y.; Liang, S.; Wang, J.; Ni, M.; Zhang, X.; Yu, F.; Chen, M.; Zhang, H.; Yan, L.; et al. Prevalence of Doravirine Cross-Resistance in HIV-Infected Adults Who Failed First-Line ART in China, 2014-18. J. Antimicrob. Chemother. 2022, 77, 1119–1124.
- Novak, R.M.; Chen, L.; MacArthur, R.D.; Baxter, J.D.; Huppler Hullsiek, K.; Peng, G.; Xiang, Y.; Henely, C.; Schmetter, B.; Uy, J.; et al. Prevalence of Antiretroviral Drug Resistance Mutations in Chronically HIV-Infected, Treatment-Naive Patients: Implications for Routine Resistance Screening before Initiation of Antiretroviral Therapy. Clin. Infect. Dis. 2005, 40, 468–474.
- Wensing, A.M.J.; van de Vijver, D.A.; Angarano, G.; Asjö, B.; Balotta, C.; Boeri, E.; Camacho, R.; Chaix, M.-L.; Costagliola, D.; De Luca, A.; et al. Prevalence of Drug-Resistant HIV-1 Variants in Untreated Individuals in Europe: Implications for Clinical Management. J. Infect. Dis. 2005, 192, 958–966.
- Brenner, B.G.; Coutsinos, D. The K65R Mutation in HIV-1 Reverse Transcriptase: Genetic Barriers, Resistance Profile and Clinical Implications. HIV 2009, 3, 583–594.
- Ross, L.; Elion, R.; Lanier, R.; Dejesus, E.; Cohen, C.; Redfield, R.R.; Gathe, J.C.; Hsu, R.K.; Yau, L.; Paulsen, D.; et al. Modulation of K65R Selection by Zidovudine Inclusion: Analysis of HIV Resistance Selection in Subjects with Virologic Failure Receiving Once-Daily Abacavir/Lamivudine/Zidovudine and Tenofovir DF (Study COL40263). AIDS Res. Hum. Retrovir. 2009, 25, 665–672.
- Ren, J.; Stammers, D.K. Structural Basis for Drug Resistance Mechanisms for Non-Nucleoside Inhibitors of HIV Reverse Transcriptase. Virus Res. 2008, 134, 157–170.
- Antinori, A.; Zaccarelli, M.; Cingolani, A.; Forbici, F.; Rizzo, M.G.; Trotta, M.P.; Di Giambenedetto, S.; Narciso, P.; Ammassari, A.; Girardi, E.; et al. Cross-Resistance among Nonnucleoside Reverse Transcriptase Inhibitors Limits Recycling Efavirenz after Nevirapine Failure. AIDS Res. Hum. Retrovir. 2002, 18, 835–838.
- Ruxrungtham, K.; Pedro, R.J.; Latiff, G.H.; Conradie, F.; Domingo, P.; Lupo, S.; Pumpradit, W.; Vingerhoets, J.H.; Peeters, M.; Peeters, I.; et al. Impact of Reverse Transcriptase Resistance on the Efficacy of TMC125 (Etravirine) with Two Nucleoside Reverse Transcriptase Inhibitors in Protease Inhibitor-Naïve, Nonnucleoside Reverse Transcriptase Inhibitor-Experienced Patients: Study TMC125-C227. HIV Med. 2008, 9, 883–896.
- Varghese, V.; Shahriar, R.; Rhee, S.-Y.; Liu, T.; Simen, B.B.; Egholm, M.; Hanczaruk, B.; Blake, L.A.; Gharizadeh, B.; Babrzadeh, F.; et al. Minority Variants Associated with Transmitted and Acquired HIV-1 Nonnucleoside Reverse Transcriptase Inhibitor Resistance: Implications for the Use of Second-Generation Nonnucleoside Reverse Transcriptase Inhibitors. J. Acquir. Immune Defic. Syndr. 2009, 52, 309–315.
- Lambert-Niclot, S.; Charpentier, C.; Storto, A.; Fofana, D.; Soulie, C.; Fourati, S.; Wirden, M.; Morand-Joubert, L.; Masquelier, B.; Flandre, P.; et al. Rilpivirine, Emtricitabine and Tenofovir Resistance in HIV-1-Infected Rilpivirine-Naive Patients Failing Antiretroviral Therapy. J. Antimicrob. Chemother. 2014, 69, 1086–1089.
- Melikian, G.L.; Rhee, S.-Y.; Varghese, V.; Porter, D.; White, K.; Taylor, J.; Towner, W.; Troia, P.; Burack, J.; Dejesus, E.; et al. Non-Nucleoside Reverse Transcriptase Inhibitor (NNRTI) Cross-Resistance: Implications for Preclinical Evaluation of Novel NNRTIs and Clinical Genotypic Resistance Testing. J. Antimicrob. Chemother. 2014, 69, 12–20.
- Brenner, B.; Turner, D.; Oliveira, M.; Moisi, D.; Detorio, M.; Carobene, M.; Marlink, R.G.; Schapiro, J.; Roger, M.; Wainberg, M.A. A V106M Mutation in HIV-1 Clade C Viruses Exposed to Efavirenz Confers Cross-Resistance to Non-Nucleoside Reverse Transcriptase Inhibitors. AIDS 2003, 17, F1–F5.
- Ali, A.; Bandaranayake, R.M.; Cai, Y.; King, N.M.; Kolli, M.; Mittal, S.; Murzycki, J.F.; Nalam, M.N.L.; Nalivaika, E.A.; Özen, A.; et al. Molecular Basis for Drug Resistance in HIV-1 Protease. Viruses 2010, 2, 2509–2535.
- Munerato, P.; Sucupira, M.C.; Oliveros, M.P.R.; Janini, L.M.; de Souza, D.F.; Pereira, A.A.; Inocencio, L.A.; Diaz, R.S. HIV Type 1 Antiretroviral Resistance Mutations in Subtypes B, C, and F in the City of São Paulo, Brazil. AIDS Res. Hum. Retrovir. 2010, 26, 265–273.
- Blanco, J.-L.; Varghese, V.; Rhee, S.-Y.; Gatell, J.M.; Shafer, R.W. HIV-1 Integrase Inhibitor Resistance and Its Clinical Implications. J. Infect. Dis. 2011, 203, 1204–1214.
- Geretti, A.M.; Armenia, D.; Ceccherini-Silberstein, F. Emerging Patterns and Implications of HIV-1 Integrase Inhibitor Resistance. Curr. Opin. Infect. Dis. 2012, 25, 677–686.
- Clutter, D.S.; Jordan, M.R.; Bertagnolio, S.; Shafer, R.W. HIV-1 Drug Resistance and Resistance Testing. Infect. Genet. Evol. 2016, 46, 292–307.
- Cooper, D.A.; Steigbigel, R.T.; Gatell, J.M.; Rockstroh, J.K.; Katlama, C.; Yeni, P.; Lazzarin, A.; Clotet, B.; Kumar, P.N.; Eron, J.E.; et al. Subgroup and Resistance Analyses of Raltegravir for Resistant HIV-1 Infection. N. Engl. J. Med. 2008, 359, 355–365.
- Lennox, J.L.; DeJesus, E.; Lazzarin, A.; Pollard, R.B.; Madruga, J.V.R.; Berger, D.S.; Zhao, J.; Xu, X.; Williams-Diaz, A.; Rodgers, A.J.; et al. Safety and Efficacy of Raltegravir-Based versus Efavirenz-Based Combination Therapy in Treatment-Naive Patients with HIV-1 Infection: A Multicentre, Double-Blind Randomised Controlled Trial. Lancet 2009, 374, 796–806.
- Han, Y.-S.; Mesplède, T.; Wainberg, M.A. Differences among HIV-1 Subtypes in Drug Resistance against Integrase Inhibitors. Infect. Genet. Evol. 2016, 46, 286–291.
- Trkola, A.; Kuhmann, S.E.; Strizki, J.M.; Maxwell, E.; Ketas, T.; Morgan, T.; Pugach, P.; Xu, S.; Wojcik, L.; Tagat, J.; et al. HIV-1 Escape from a Small Molecule, CCR5-Specific Entry Inhibitor Does Not Involve CXCR4 Use. Proc. Natl. Acad. Sci. USA 2002, 99, 395–400.
- Moore, J.P.; Kuritzkes, D.R. A Pièce de Resistance: How HIV-1 Escapes Small Molecule CCR5 Inhibitors. Curr. Opin. HIV AIDS 2009, 4, 118–124.
- Lalezari, J.P.; Eron, J.J.; Carlson, M.; Cohen, C.; DeJesus, E.; Arduino, R.C.; Gallant, J.E.; Volberding, P.; Murphy, R.L.; Valentine, F.; et al. A Phase II Clinical Study of the Long-Term Safety and Antiviral Activity of Enfuvirtide-Based Antiretroviral Therapy. AIDS 2003, 17, 691–698.
- Cabrera, C.; Marfil, S.; García, E.; Martinez-Picado, J.; Bonjoch, A.; Bofill, M.; Moreno, S.; Ribera, E.; Domingo, P.; Clotet, B.; et al. Genetic Evolution of Gp41 Reveals a Highly Exclusive Relationship between Codons 36, 38 and 43 in Gp41 under Long-Term Enfuvirtide-Containing Salvage Regimen. AIDS 2006, 20, 2075–2080.
- Lu, J.; Deeks, S.G.; Hoh, R.; Beatty, G.; Kuritzkes, B.A.; Martin, J.N.; Kuritzkes, D.R. Rapid Emergence of Enfuvirtide Resistance in HIV-1-Infected Patients: Results of a Clonal Analysis. J. Acquir. Immune Defic. Syndr. 2006, 43, 60–64.
- Marcelin, A.-G.; Reynes, J.; Yerly, S.; Ktorza, N.; Segondy, M.; Piot, J.-C.; Delfraissy, J.-F.; Kaiser, L.; Perrin, L.; Katlama, C.; et al. Characterization of Genotypic Determinants in HR-1 and HR-2 Gp41 Domains in Individuals with Persistent HIV Viraemia under T-20. AIDS 2004, 18, 1340–1342.
- Menzo, S.; Castagna, A.; Monachetti, A.; Hasson, H.; Danise, A.; Carini, E.; Bagnarelli, P.; Lazzarin, A.; Clementi, M. Genotype and Phenotype Patterns of Human Immunodeficiency Virus Type 1 Resistance to Enfuvirtide during Long-Term Treatment. Antimicrob. Agents Chemother. 2004, 48, 3253–3259.
- Sista, P.R.; Melby, T.; Davison, D.; Jin, L.; Mosier, S.; Mink, M.; Nelson, E.L.; DeMasi, R.; Cammack, N.; Salgo, M.P.; et al. Characterization of Determinants of Genotypic and Phenotypic Resistance to Enfuvirtide in Baseline and On-Treatment HIV-1 Isolates. AIDS 2004, 18, 1787–1794.
- Chang, L.; Zhao, J.; Guo, F.; Ji, H.; Zhang, L.; Jiang, X.; Wang, L. HIV-1 Gp41 Genetic Diversity and Enfuvirtide Resistance-Associated Mutations among Enfuvirtide-Naïve Patients in Southern China. Virus Res. 2021, 292, 198215.
- Kageyama, S.; Amolong Hinay, A.; Telan, E.F.O.; Samonte, G.M.J.; Leano, P.S.A.; Tsuneki-Tokunaga, A.; Kanai, K. Intrinsic Replication Competences of HIV Strains After Zidovudine/Lamivudine/Nevirapine Treatment in the Philippines. J. Int. Assoc. Provid. AIDS Care 2019, 18, 2325958219856579.