Two years later, in 1985, HIV-2 was identified as partially responsible for the AIDS pandemic but causing a milder form of immunodeficiency; data about HIV-2 are still limited, and only a few subtypes have been described.
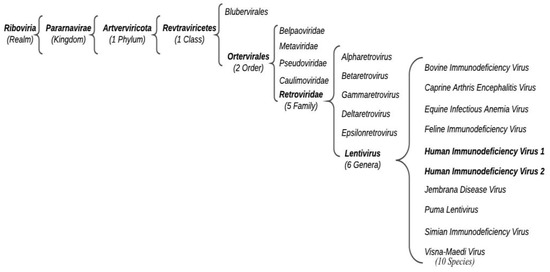
According to the International Committee for the Taxonomy of Viruses (ICTV), HIV belongs to the Retroviridae family, Orthoretrovirinae subfamily, and Lentivirus genus (Figure 1).
Based on different genome sequences risen from an independent cross-species transmission event, thwe researchers ccan distinguish HIV-1 groups M (major), O (outlier), N (non-M, non-O), and P.
3. Treatment
Before the discovery of antiretroviral therapy (ART), HIV/AIDS patients were initially treated with only one or two antiretroviral drugs, which belonged to the class of nucleotide analogs, whose abilities to reduce viral load were very low and hampered by the rapid insurgence of resistance and, consequently, were not able to modify the course of the disease. Approximately 23 years ago, ART, which is based on the combination of different classes of antiretroviral drugs, was developed. This treatment has significantly prolonged survival in patients with AIDS (about 7 to 10 years of survival or longer). The first drug to treat HIV/AIDS approved by the US Food and Drug Administration was zidovudine, a nucleoside reverse-transcriptase inhibitor (NRTI), in 1987, but studies highlighting the effectiveness of combined administration of two drugs NRTI were already released at the beginning of 1990
[26][27][28][26,27,28].
ART can include a combination of nucleoside reverse-transcriptase inhibitors (NRTIs), non-nucleoside reverse-transcriptase inhibitors (NNRTIs), protease inhibitors (PIs), integrase strand transfer inhibitors (INSTIs), and entry inhibitors.
Since these drugs belong to different classes, they have different mechanisms of action by which they interfere with a unique step in the life cycle of the virus. Generally, two NRTIs are used in association with another class, such as PI or INSTI, but current data highlight that a simplified two-drug treatment can be equally effective
[29]. All these treatments can be modified based on toxicity and induced resistance.
NNRTIs and NRTIs inhibit the polymerase activities of HIV-1 reverse transcriptase (RT), which plays a pivotal role in the HIV life cycle. Reverse transcriptase (RT) is essential for the life cycle of HIV because it converts ingle-stranded genomic RNA into double-stranded DNA, which is subsequently integrated into the host chromosome and passed on to all progeny cells
[30].
For that reason, HIV RT has been an ideal target for antiretroviral agents. NRTI drugs approved and used in ART are abacavir, didanosine, emtricitabine, lamivudine, stavudine, tenofovir, and zidovudine, while NNRTI drugs are doravirin, delavirdine, efavirenz, etravirine, nevirapine, and rilpivirine. Many of these drugs are not suggested by current guidelines, owing to several considerations regarding their toxicity or antiviral effects.
Protease inhibitors (PIs) are a major class of drugs effective in treating HIV infection/AIDS because of their high effectiveness in inhibiting HIV replication and their relatively low resistance rate. In the past several years, a variety of protease inhibitors have been found
[31]. As expected, these drugs interact with HIV-1 protease, which is responsible for the production of all viral enzymes and structural proteins necessary to produce mature, virulent virions, starting from long polypeptide chains, which are cut to obtain single proteins
[32][33][32,33].
Therefore, the inhibition of the protease activity effectively interferes with a vital stage in the HIV life cycle
[34]. PI drugs include atazanavir, darunavir, fosamprenavir, indinavir, nelfinavir, ritonavir, saquinavir, and tipranavir; presently, darunavir is the most frequently used.
The integrase (IN) is an enzyme that HIV uses to insert its viral DNA (vDNA) into the DNA of the host cell through two catalytic actions: 3′ processing and strand transfer.
Although the integrase strand transfer inhibitor (INSTI) antiretroviral drugs are the latest class of agents approved
[34][35][34,35], they have a key role in the treatment of an HIV-positive individual by inhibiting the strand transfer step made by INs through competitive binding to the enzyme’s active site. Then, INSTIs stop IN action also by chelating the divalent cation (Mg
2+ or Mn
2+) that is required for its enzymatic activity
[36].
There are currently four INSTIs approved for the treatment of HIV infection: raltegravir, elvitegravir, bictegravir, and dolutegravir
[37].
Finally, entry inhibitors prevent the virus from entering the cell and include CCR5 antagonist and entry inhibitor of gp41. HIV-1 uses various membrane proteins as a receptor to bind the plasma membrane, such as CCR5
[38][39][38,39], or uses gp41, a glycoprotein that physically brings the viral envelope closer to that of the cell membrane, allowing their fusion. Maraviroc binds CCR5, while enfuvirtide binds gp41
[40], preventing the virus’s cell binding or induced membrane fusion and, consequently, its entry into the cell.
Other noteworthy and recent drug classes are capsid inhibitors and NNRTIs.
Lenacapavir represents a new class of drugs called capsid inhibitors
[41], approved in December 2022 for the first time by the FDA. Its mechanism of action is inhibiting the disassembly of the capsid shell, a step essential for viral replication. It is used in association with other drugs.
NNRTIs, such as islatravir, which is still in phase 3 clinical trial, inhibit reverse transcription via multiple mechanisms, which can confer a unique mutant susceptibility profile
[42].
The treatment for HIV-2 is not the same as the one for HIV-1; in fact, drugs used to treat HIV-2 are NNRTIs and entry inhibitors, while some PIs have weak or no inhibitory activity against HIV-2
[17].
Some people receiving ART have shown severe side effects, such as diarrhea, headache, fatigue, rash, or, worse, treatment failure. Nevertheless, ART has been revealed to be really successful and has allowed the majority of infected people to conduct their normal lives.
The emergence of HIV drug-resistant subtypes, especially caused by the rapid and error-prone replication of the virus, is increasing
[30].
Moreover, along with a growing number of drug-resistant strains, ART has led to an ever-growing pool of individuals who can transmit drug-resistant strains of HIV-1
[43][44][43,44].
4. Drug Resistance Mechanism
HIV-1 virus has a high mutation rate, accumulating nearly one nucleotide mutation per replication cycle
(Table S1). That is the main reason why there is a considerable HIV-1 variation in patients
[45][46][45,46].
Since people are usually infected with only a single or a few original clones
[47], an exponential number of virions are likely produced each day in untreated individuals, resulting in innumerable variants of the virus. The high HIV-1 recombination rate, due to different variants infecting the same cell, together with its capacity of reactivation in infected cells, increases the complexity of this virus
[48][49][48,49]. As a matter of fact, it is possible to find a wide spectrum of virus variants infecting the same patient. Unfortunately, most of the time, these variants gain new mechanisms to evade the immune system of the host
[50].
People harboring HIV-resistant strains can be divided into two categories: people on ART that acquire HIV drug resistance (ADR) and people who become infected with HIV-resistant strains.
Among people receiving ART, resistance can frequently develop, prompted by suboptimal drug treatment, which can lead to the selection of drug-resistant viruses
[46]. When virus replication occurs in the presence of suboptimal concentrations of the drug, drug-resistant viruses are selected, and the replication of drug-resistant viruses in the presence of the drug can further increase the virus’ mutation rate
[46].
Other than acquired drug resistance and transmitted resistance, naturally drug-resistant viruses are extremely rare when the viruses are not subjected to selective drug pressure, even more in untreated patients. HIV-1 genetic variability is ensued by the HIV-1 reverse transcriptase (RT) processing errors by recombination when more than one viral variant infects the same cell and by the accumulation of proviral variants during the course of infection
[45][49][51][52][45,49,51,52]. Although most HIV-1 infections are initiated by a single viral variant
[39], innumerable variants related to the initially transmitted virus emerge within weeks following infection
[51][53][51,53].
There are often high levels of cross-resistance within each drug class. Most drug resistance mutations in a specific antiretroviral class decrease susceptibility to one or more antiretroviral drugs of the same class
[54]. In contrast, viruses with high levels of drug resistance in one specific antiretroviral class are generally susceptible to drugs that belong to another class. However, there are few cases of cross-resistance between drug classes. Recently, Sun et al. demonstrated the prevalence of doravirine cross-resistance in HIV-infected adults who failed first-line ART
[55]. Doravirine resistance was highly associated with efavirenz and nevirapine resistance and moderately with etravirine and rilpivirine resistance.
Several studies have recently shown an increase in primary resistance rates in various geographic areas
[56][57][56,57], and a genotypic resistance test is now recommended before the initiation of ART in patients with HIV infection.
5. NRTI Drug Resistance
Nucleoside reverse-transcriptase inhibitors (NRTIs) form the backbone of ART
[58].
NRTI drug resistance works in two different biochemical ways. The first mechanism relates to the RT enzyme’s ability to avoid the binding of the NRTI, while retaining the ability to recognize the natural dNTP substrates during polymerization. The second mechanism, instead, relates to the promotion of the hydrolytic removal of the chain terminating NRTI, continuing DNA synthesis
[54].
Therefore, HIV has two major routes to escape nucleoside and nucleotide reverse-transcriptase inhibitor class drug selection pressure. One path is through the selection of thymidine analog mutations (TAMs; M41L, D67N, K70R, L210W, T215F/Y, and K219Q = E), which act to increase excision of NRTIs. Interestingly, the TAM pathway 1, which includes mutations at codons 41, 210, and 215, was prevalent among subtype B viruses, whereas mutations at codons 67, 70, and 219 were the most present among subtype C and F viruses. These mutations can be selected either by zidovudine (ZDV) or by stavudine and confer resistance to these drugs
[30][59][30,59].
6. NNRTIs Drugs Resistance
NNRTIs have a relatively low genetic barrier to resistance. In general, NNRTIs interact with a hydrophobic pocket within the HIV-1 reverse-transcriptase enzyme. Almost all NNRTI-resistance mutations are located in the RT domain hosting this binding pocket
[60][73]. Since the NNRTI-resistance mutation reduces susceptibility to two or more NNRTIs and there are multiple independent NNRTI-resistant lineages, because of the low genetic barrier, there is a high level of cross-resistance inside this drug’s class
[54][61][62][63][54,74,75,76].
As previously described, there is a significant difference between B and non-B subtype viruses. Moreover, non-B subtype viruses were found to have more resistance-associated mutations than B subtype viruses. This difference is mainly due to the widespread mutation E138A, which is twice as common among non-B subtype viruses than B subtype viruses
[64][67].
However, the most common NNRTI mutations are L100I, K101E/P, K103N/S, V106A/M, Y181C/I/V, Y188C/H/L, G190A/S/E, and M230L. K103N is the most frequent NNRT-associated mutation. Each of these mutations, except for L100I, causes intermediate- or high-level phenotypic resistance to nevirapine and, with the exception of Y181C/I/V, efavirenz
[65][77]. It has also been observed that they all cause phenotypic etravirine and rilpivirine resistance except for K103N/S, V106A/M, Y188C/H/L, and G190A/S/E64.
The NNRTI-resistance mutation V106M occurs more often in subtype C viruses from patients treated with nevirapine or efavirenz because V106M requires a single base-pair change in subtype C viruses—GTG (V) => ATG (M)—but a two base-pair change in all other subtypes—GTA (V) => ATG (M)
[66][78].
7. PIs Drugs Resistance
PIs have high genetic barriers to resistance; they act as competitive inhibitors in the active site of HIV-1 protease that usually prevents the enzyme from processing the Gag and Gag/Pol polyprotein precursors necessary for viral maturation
[67][90].
In subtype B viruses, resistance mutations at codons 33, 34, 58, 63, 73, 71, 77, and 84 are more common than in subtype F or C viruses, whereas those at codons 20, 36, and 89 are less common. In subtype F viruses, resistance mutations are more common at codons 10, 20, 35, 36, 48, 74, 57, 82, and 89, whereas they are less common at codons 47 and 93. In subtype C viruses, the frequency of resistance mutations is higher at codons 20, 36, 89, and 93 and lower at codons 10, 30, 43, 46, and 74
[68][91].
PI-resistance mutations are different: there are major ones and accessory ones, which both act by reducing susceptibility to one or more PIs. The accessory PI-resistance mutations, however, act in combination with the major PI-resistance mutations
[54].
8. INSTIs Drugs Resistance
INSTIs block the action of the integrase viral enzyme (responsible for the insertion of the HIV-1 genome into the host DNA) by impeding the correct positioning of viral DNA at the active site of the enzyme and by binding to the catalytic metal cations inside the retroviral IN active site
[54]. Raltegravir resistance occurs by three main, occasionally overlapping, mutational pathways: N155H ± E92Q; Q148H/R/K ± G140S/A; Y143C/R
[69][70][108,109]. Each of these pairs of mutations is often accompanied by other accessory mutations
[69][108]. N155H, Q148R, Y143R, and other mutations have been shown to reduce raltegravir susceptibility. It is very important to consider that except for Y143C/R, most raltegravir-resistance mutations confer cross-resistance to elvitegravir.
Likewise, most elvitegravir-resistance mutations are the cause of raltegravir cross-resistance. Dolutegravir requires a Q148 mutation in combination with E138 ± G140 since it has a higher genetic barrier to resistance than raltegravir and elvitegravir, requiring a Q148 mutation in combination with E138 ± G140
[71][85]. However, there are other mutations, including N155H in combination with Q148, that appear to increase the risk of developing resistance to dolutegravir.
The fact that a single mutation can reduce raltegravir (and elvitegravir) susceptibility more than 10-fold suggests that these inhibitors have a low genetic barrier to resistance
[72][73][110,111].
Significant variability in INSTI-associated mutations can influence integrase activity and viral resistance among HIV-1 subtypes
[74][112].
9. Entry Inhibitor Drugs Resistance
9.1. Chemokine Receptor 5 (CCR5) Antagonists
The small-molecule inhibitor maraviroc allosterically inhibits the binding of HIV-1 gp120 to the host CCR5 (R5) co-receptor. Its use is limited to the early phases of HIV infection because CCR5 tropic virus prevalence is very low in the advanced phase of the infection. Mutations seen in patients enrolled in the MOTIVATE trials include G11S + I26V, S18G + A22T, A19S + I26V, I20F + A25D + I26V, and I20F + Y21I in the V3 region. Mutation-conferring resistance to maraviroc affects the V3 loop region and involves mutations that result in increased affinity of gp120 to MVC-bound CCR5, enabling gp120 binding to CCR5 despite conformational changes from MVC binding
[75][113]. Studies on maraviroc resistance patterns are limited
[75][76][113,114].
9.2. Entry Inhibitor of gp41
Enfuvirtide, the only approved entry inhibitor, binds to gp41, preventing the creation of the entry pore for the capsid of the virus and keeping it out of the cell
[77][115].
Enfuvirtide has a low genetic barrier
[78][79][116,117]. Mutations that confer enfuvirtide resistance are localized in its binding site, gp41, in codons 36–45
[80][81][82][118,119,120]. In particular, when only one single mutation affects the site of the enfuvirtide, susceptibility is reduced by about 10-fold, while two mutations lead to a reduction of about 100-fold. The most common enfuvirtide mutations are G36DEV, V38EA, Q40H, N42T, and N43D
[71][85]. However, in a recent study, various polymorphisms were observed in the envelope gp41 region. The major polymorphisms were R46K/M/Q, E137K, and S138A. R46K/M/Q and S138A were predominant in subtype CRF07_BC, and E137K was prevalent in subtype B
[83][121].
10. Conclusions
HIV drug resistance is present all over the world. Approximately 38 million people across the globe live with HIV/AIDS, and of these, 36.2 million are adults, and 1.8 million are children (<15 years old). The different available studies concerning the emergence of HIV drug resistance are still in their infancy but pave the way to help in understanding the possible future steps to follow.
TheOur work tried to highlight the large number of mutations present globally and underline that many drugs previously administered as part of ART during recent years have lost their efficacy, owing to viral resistance development, prompting new studies in this field to identify new drugs using other mechanisms to inhibit HIV replication.
ART has been shown to reduce HIV mortality, morbidity, and transmission. However, the presence of HIV subtypes with increasing drug resistance can compromise the effectiveness of antiretroviral drugs
[71][85]. The different subtypes of HIV-1 and HIV-2 predominate in certain geographic areas, but their distribution is becoming increasingly heterogeneous as the pandemic progresses. As shown above, certain subtypes, in particular the resistant ones, may be associated with an increased risk of transmission and a faster progression to the development of AIDS.
The rise in HIV drug resistance is one of the greatest threats to global health that can result in millions of deaths. Diagnostic tools such as resistance testing should be considered an integral part of the plan to address this problem. Nevertheless, it is important to remember the limits of these tests since they only report mutations already known to be associated with drug resistance, while they are less interpretable when compared to new drugs or newer mutations. Plus, the interpretation of their results requires a wide expert panel. However, such testing, based on NGS sequencing, can be helpful in estimating the intrinsic replication competence of the virus
[84][122], but resistance testing is currently only recommended for drug-experienced patients for whom therapy is not successful. Resistance testing should, instead, be applied to treatment-naïve patients to find any mutation resistance before beginning the treatment.
Although great progress has been made in monitoring population-level emergence and transmission of HIV drug resistance mutations, the mutated and resistant strains that reemerge under drug-induced selective pressure need to be effectively detected and preferentially extinguished to prevent the development of AIDS.