Skin cancer is one of the most common types of cancer, and its incidence continues to increase. It is divided into two main categories, melanoma and non-melanoma. Treatments include surgery, radiation therapy, and chemotherapy. The relatively high mortality in melanoma and the existing recurrence rates, both for melanoma and non-melanoma, create the need for studying and developing new approaches for skin cancer management. Studies have focused on immunotherapy, photodynamic therapy, photothermal therapy, and photoimmunotherapy. Photoimmunotherapy has gained much attention due to its excellent potential outcomes. It combines the advantages of photodynamic and/or photothermal therapy with a systemic immune response, making it ideal for metastatic cancer.
- immunotherapy
- photothermal therapy
- photodynamic therapy
- melanoma
- basal cell carcinoma
- squamous cell carcinoma
1. Introduction
1.1. Immunotherapy
Cancer immunotherapy has been contributing to improved survival and quality of life for cancer patients. It consists in triggering the immune system to control and fight cancer, overcoming the mechanisms that cancer cells develop to escape immune surveillance and avoid detection and elimination [18][19][18,19]. Immunotherapy started more than 100 years ago, in New York, with Dr. Coley, who treated sarcoma patients with Coley’s toxin, a vaccine with a mixture of two bacterial toxins [20]. Currently, there are two types of immunotherapies, active and passive immunotherapy. Active immunotherapy stimulates the patient’s immune system, whereas passive immunotherapy can be with the administration of, for example, cytokines, vaccines, and antibodies [21][22][23][21,22,23]. The immune system plays a critical role in recognizing, eliminating, and controlling tumor progression. However, cancer cells develop mechanisms to avoid it, namely: (i) downmodulation of components of antigen processing and presentation machinery; (ii) an environment that promotes suppressor immune cells, such as regulatory T cells (Treg), an immunosuppressive subset of CD4+ T-cell family, myeloid-derived suppressor cells (MDSCs), and tumor-associated macrophages, which are anti-inflammatory macrophages (M2- like); (iii) production of soluble factors associated with immunosuppression, such as TGF-β and IL-10; (iv) and upregulation of ligands for coinhibitory receptors that downmodulate programmed death ligand-1 (PD-L1) [24][25][26][27][28][29][24,25,26,27,28,29]. Dendritic cells (DCs) induce the differentiation of T cells to their antigen-specific effector T cells. CD4+ T cells are responsible for inducing DC maturation and for CD8+ T-cell priming. The primed cells are activated to form cytotoxic T lymphocytes, and these are responsible for releasing INF-γ and TNF-α, which will induce cytotoxicity in cancer cells. INF-γ is produced by both CD4+ and CD8+ T cells and stimulates the antitumor pro-inflammatory macrophages (M1). These tumor suppressor cells, such as cytotoxic T lymphocytes, also upregulate the release of pro-inflammatory cytokines, namely, IL-2, IL-6, IL-12, INF-γ, and TNF-α [24][30][31][32][24,30,31,32]. The higher calreticulin (CRT) exposure and release of high mobility group box 1 (HMGB-1) also act as an “eat-me” signal to induce tumor cell apoptosis [33][34][35][36][33,34,35,36].1.2. Phototherapy for Cancer Treatment
Phototherapy (PT) started 4000 years ago in ancient Egypt to treat Vitiligo, when a plant extract was boiled and then combined with sun exposure. Modern phototherapy, on the other hand, started only in the 70s, using artificial light sources [37][38][39][37,38,39]. Phototherapy is mainly divided into two categories, photodynamic therapy (PDT) and photothermal therapy (PTT). In PDT, a photosensitizer agent is irradiated by light to generate reactive oxygen species (ROS). These are highly toxic, causing cell death. PTT is based on local temperature increase, usually triggered by laser radiation. Usually, NIR lasers (650–1350 nm) are used in PTT due to their efficiency in penetrating tumors [40][41][42][43][40,41,42,43]. PTT can be divided into two categories. In the first, called mild hyperthermia, temperature increases up to 43–50 °C, leading to enhanced membrane permeability, cellular uptake, metabolic signaling disruption, and dysfunctional membrane transport. The capability of tumor cells to recover from such damages is very low. The second one is photothermal ablation (>50 °C), which destroys the cellular membrane, leading to necrotic cell death [44]. PDT has been FDA-approved for almost 40 years [40][45][40,45]. Hematopotphyrin derivative (HPD) was the first PS receiving FDA approval, nowadays Foscan®, Levulan®, Radachlorin®, Metvix®, and Photofrin® are FDA-approved PS [39][43][39,43].1.3. Photoimmunotherapy for Cancer Treatement
PT triggers immunogenic cell death (ICD) that will release tumor-specific antigens (TSAs) and damage-associated molecular patterns (DAMPs), namely CRT, HMGB-1, and ATP. This phenomenon increases the immunogenicity of the tumor microenvironment once DAMPs induce the maturation of DCs, and pro-inflammatory cytokines, such as IL-2, IL-6, IL-12, INF-γ and TNF-α, were also reported to increase. The immunostimulatory effect of PT boosts anti-tumor immunity when compared to immunotherapy alone. Although immunotherapy by itself can be effective in triggering the immune response at tumor site, it is inefficient to eradicate primary tumors [46][47][48][49][50][46,47,48,49,50]. When combining phototherapy with immunotherapy, in photoimmunotherapy (PIT), a synergy is reported to occur between them. PT directly kills the tumor cells and triggers a systemic immune response, and when in combination with immunotherapy, immunological memory is formed. Photoimmunotherapy has the advantages of phototherapy and the ability to trigger an immune response, making it ideal for treating metastatic cancer. Thereby, PIT eradicates primary tumors and, through simultaneously stimulating immune memory, it has the potential to prevent tumor recurrence and metastasis [42][46][51][52][42,46,51,52].2. Nanomaterials
2.1. Structure and Properties
In recent years, nanomaterials have gained much interest in the biomedical field, namely in drug delivery, tissue engineering, diagnosis, and theragnostics, amongst others. Nanomaterials can be divided into different categories according to their properties (e.g., size, shape, physicochemical properties, etc.). Regarding nanomaterials used for skin cancer photoimmunotherapy, the focus of this resviearchw, the main categories found are metallic, polymeric, lipid-based and 2D nanomaterials [53], as illustrated in Figure 1.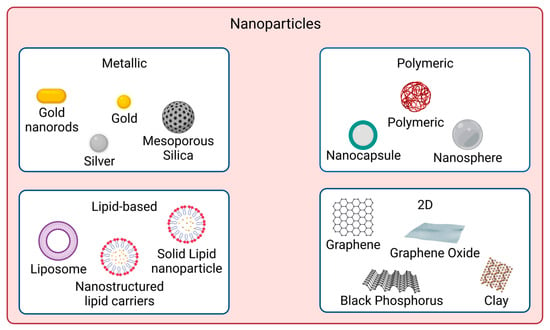
2.2. Surface Modification and Encapsulation Strategies
Nanomaterials to be used for biomedical applications should have good stability in physiological conditions and biocompatibility. Many nanoparticles agglomerate in aqueous solutions due to being hydrophobic or creating strong interparticle interactions, for instance. Covalent and non-covalent surface functionalization can be performed to enhance nanoparticles properties. However, such modifications should not affect nanomaterials photoabsorption properties if skin cancer photoimmunotherapy applications are desired. Figure 2 shows examples of such approaches.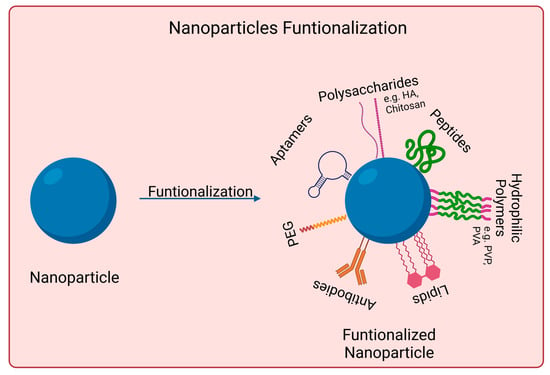
3. Skin Cancer Photoimmunotherapy Studies
Skin cancer is one of the most common cancers. Although there are already several treatment options available, there is still an urgent need to reduce mortality, recurrence rates, and side effects [3][8][91][3,8,91]. Photoimmunotherapy has gained much interest in recent years. It combines the advantages of phototherapy with enhancement of the immune response, resulting in a more effective cancer treatment approach. Figure 3 shows the mechanisms of action of photoimmunotherapy. Table 1 summarizes the state-of-the-art literature regarding nanomaterials used for skin cancer photoimmunotherapy, including their composition, size, and biological effects.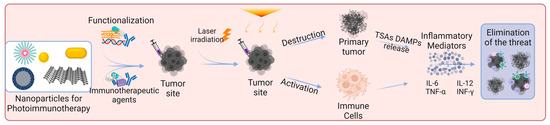
|
Nanomaterial |
Physicochemical Modifications |
Loaded Substance |
Particle Size |
In Vitro Studies |
In Vivo Studies |
Ref. |
||
|---|---|---|---|---|---|---|---|---|
|
Parameters |
Results |
Parameters |
Results |
|||||
|
Aluminum hydroxide |
BSA surface adsorption |
Chlorin e6 |
25.25 ± 2.1 nm |
B16F10 cells [Al-BSA-Ce6] = 0.1 μg mL−1 I: 660 nm, 0.8 W cm−2, 5 min |
95% cell death (MTT assay) ↑CD80 ↑TNF-α, IL-12p70, IL-1β |
C57BL/6 mice subcutaneously injected with B16F10 cells [Al-BSA-Ce6] = 5 mg kg−1 I: 660 nm, 0.8 W cm−2, 5 min |
Tumor volume of 0 mm3 at day 7 ≈63% mice survived 100 days ↑T cells tumor infiltration ↑TNF-α and INF-γ production |
[92] |
|
Black Phosphorus |
PEG electrostatic adsorption |
Imiquimod |
≈120 nm |
B16 cells [BP-PEG] = 10 μg mL−1 I: 808 nm, 3.2 W cm−2, 10 min |
45% cell viability↓ (MTT assay) ↑TNF-α, IL-6, IL-12 DCs↑ 30.8% |
C57BL/6 mice subcutaneously injected with B16 [BP] = 0.5 mg kg−1, 25 μL [R837] = 0.35 mg kg−1, 25 μL I: 808 nm, 3.2 W cm−2, 3 min |
≈10-fold tumor vol.↓ DCs↑ 45.5% ↑TNF-α, IL-6, IL-12 |
[93] |
|
Black Phosphorous |
N/A |
FKF-OVAp |
≈500 × 23 nm |
N/A |
N/A |
C57BL/6 mice subcutaneously injected with B16-OVA [FKF-OVAp] = 10 nmol per mouse [BPs] = 15.9 μg per mouse I: 808 nm, 0.5 W cm−2, 5 min |
≈3-fold tumor vol.↓ 100% survival over 60 days ↑DC activation ↑CD8+ T cells effector and central memory |
[94] |
|
Chitosan |
Cross-linking, sodium tripolyphosphate |
IDO |
220 nm |
B16 cells [ICG-NP] = 30 μg mL−1 I: 808 nm, 0.35 W cm−2, 5 min |
≈0% cell viability (CCK8 assay) ≈85% DC frequency |
C57BL/6 mice subcutaneously injected with B16F10 Drug loading: 4 and 35 μg per microneedle patch of ICG and 1-MT, respectively I: 808 nm, 0.35 W cm−2, 5 min 3 cycles at interval of 2 days |
Tumor volume of ≈0 mm3 80% survival rate without recurrence after 120 days ≈55% DC maturation level ↑CD8+ T cells in distant tumor ↑TNF-α, IL-12p70, IL-6 |
[95] |
|
Gold |
HA surface adsorption |
M2pep |
64.6 nm |
B16F10 cells [HA-AuNR/M-M2pep] = 20 μg mL−1 I: 808 nm, 1.5 W cm−2, 2 min |
40% cell viability (CCK8 assay) 35.1 ± 1.8% apoptosis (Annecin V-FITC) ↑CRT-positive cells and HMGB1 release |
C57BL/6 mice subcutaneously injected with B16F10 [AuNR] + [M2pep] = 10 + 12 mg kg−1 I: 808 nm, 1.5 W cm−2, 2 min |
≈10-fold tumor vol.↓ 67% survival rate at 45 days 3.7-fold↑ CD8+ T cells INF-γ, TNF-α↑ ≈ 6-fold |
[96] |
|
Gold |
BSA surface adsorption |
R837 |
122.1 ± 11.6 nm |
B16-F10 cells [Au] = 11.5 μg mL−1 I: 1064 nm, 1.0 W cm−2, 10 min |
Cell viability↓ to ≈27% (MTS assay) HSP70/β-Actin release ≈ 0 |
C57BL/6 mice subcutaneously injected with B16F10 [Au] = 300 μg mL−1 I: 1064 nm, 1.0 W cm−2, 10 min |
≈10-fold tumor vol.↓ TNF-α, IL-6, IL-12 ≈ 14, 9, 3 times↑ than PBS, respectively ↑CD8+ T cells infiltration |
[97] |
|
Gold |
Gold nanoparticles retained in extracellular vesicles with tumor antigens (AuNP@B16F10) |
Tumor antigens (AuNP@B16F10) |
40 nm |
N/A |
N/A |
Murine melanoma model subcutaneously injected with AuNP@DCB16F10 [Au] = 1.35 mg kg−1 I: 808 nm, 2.0 W cm−2, 1 min Cycle: 3 times, 3 days interval |
69% tumor volume↓ 50% tumor-free mice at day 19 Distant tumor inhibition ↑CD3+ and CD8+ T cells infiltration ↑INF-γ, TNF-α, IL-6 |
[98] |
|
Gold |
N/A |
SV |
50 nm |
B16-F10 cells [Au] = 60 μg mL−1 I: 808 nm, 1.5 W cm−2, 2 min |
≈12% cell viability (MTT assay) CRT expression ≈3.5-fold↑ ↑mature DCs frequency |
B16F10-bearing C57 mice I: 808 nm, 1.5 W cm−2, 2 min |
≈10-fold tumor vol.↓ ↑DC maturation CD4+ and CD8+ T cells proliferation |
[99] |
|
Hyaluronic acid |
Self-assembly of Ce6/α-linoleic acid (L-Ce6 NAs (nano-assemblies)) Fast dissolving L-Ce6 NAs in oligo-HA and micro-molding of microneedles (tips enriched with 3 µg Ce6) |
Ce6 |
≈86 nm |
B16F10 cells [Ce6] = 400 μM I: 660 nm, 200 mW cm−2, 5 min |
CRT fluorescence ↑2-fold ATP secretion ≈ 2.5 nM ↑HMGB1 release |
C57BL/6 mice subcutaneously injected with B16F10 [Ce6] = 0.12 mg kg−1 I: 660 nm, 200 mW cm−2, 4 min |
≈3-fold tumor vol.↓ ↑CD4+ and CD8+ ≈3 and 4-fold |
[100] |
|
Liposomes |
N/A |
TRP-2 |
180.4 ± 10.2 nm |
B16F10 cells [TLipIT NPs] = 100 μg mL−1 I: 808 nm, 0.75 W cm−2, 5 min |
≈12% early apoptosis (Annexin V-FITC) 37% late apoptosis ↑TNF-α, INF-γ |
C57BL/6 mice subcutaneously injected with B16F10 [TLipIT/NEs] = 100 μg mL−1 I: 808 nm, 0.75 W cm−2, 5 min |
≈10-fold tumor vol.↓ ≈33% CD80+ and CD86+ mature DCs frequency ≈49 and ≈33% CD4+ and CD8+ T lymphocytes frequency |
[101] |
|
Micelles |
N/A |
CQ IR780 |
80−90 nm |
B16 cells [C/I-Mil] = 4 μg mL−1 I: 808 nm, 1.0 W cm−2, 5 min |
20% cell viability (CCK-8 assay) Cell membrane integrity destroyed Phagocytic index ↑3.0-fold |
C57BL/6 mice subcutaneously injected with B16F10 [C/I-Mil] + [CQ/Mil] = 4 μg mL−1 + 20 μg/patch I: 808 nm, 1.0 W cm−2, 5 min |
Primary tumor suppression: 0 mm3 50% survived at least 40 days Distant tumor volume ↓3.4-fold |
[102] |
|
Micelles |
N/A |
Imiquimod |
72.0 ± 18.0 nm |
N/A |
N/A |
C57BL mice subcutaneously injected with B16 cells [IQPM] = 5 mg kg−1 I: 808 nm, 1.5 W cm−1, 5 min |
Primary tumor suppression: 0 mm3 CD8+ and CD4+ T cells ↑9.3- and 10.3-fold 2.4-fold↓ metastasis |
[103] |
|
mPEG-Pep-IDOi/ICG NPs |
N/A |
N/A |
140 nm |
B16-F10 cells [ICG] = 20 μg mL−1 I: 808 nm, 1.0 W cm−2, 5 min |
≈0% cell viability (CCK-8 assay) Induced ICD of tumor cells ≈70% CD80 and CD86↑ |
C57BL/6 mice subcutaneously injected with B16-F10 [ICG] = 4 mg kg−1 [IDOi] = 5 mg kg−1 I: 808 nm, 1.0 W cm−2, 5 min |
Primary tumor suppression: 0 mm3 CD80+ and CD86+ ↑13.5 and 12.3% ↑INF-γ, TNF-α, IL-6 |
[104] |
|
PBE |
N/A |
RSL-3 |
<100 nm |
B16-F10 cells [RSL-3] = 0.5 μg mL−1 [INF-γ] = 100 ng mL−1 I: 671 nm, 100 mW cm−2, 1 min |
30% mature DC CTR expression ↑5.0-fold |
C57BL/6 mice subcutaneously injected with B16-F10 [RSL-3] = 0.5 μg mL−1 [INF-γ] = 100 ng mL−1 I: 671 nm, 150 mW cm−2, 2 min |
≈2-fold tumor vol.↓ ≈50% survival rate ≈30% mature DC cells INF-γ secretion ↑ 4-fold |
[105] |
|
PEI-PBA |
PEG surface adsorption |
Ce6 aPDL1 |
117 ± 4.0 nm |
B16F10 cells [NC@Ce6-pH 6.0] = 7.5 μg mL−1 I: 650 nm, 20 mW cm−2, 2.5 min |
89% CRT rate ≈34% apoptosis (Annexin V-FITC) DC maturation |
B16F10 tumor-bearing mice [Ce6] = 2.5 mg kg−1 I: 650 nm, 100 mW cm−2, 10 min |
78% tumor inhibition rate ≈49% tumor infiltrating T cells DC maturation |
[106] |
|
Polydopamine |
PEI surface adsorption |
CpG oligodeoxynucleotides |
140 nm |
B16F10 cells [PPP/CpG/HA] = 200 μg mL−1 I: 808 nm, 2.0 W cm−2, 5 min |
≈5% cell viability (MTT assay) ≈60% apoptosis (Annexin V-FITC) ≈60% CD80+ DC ≈50% CD86+ DC |
C57BL/6 mice subcutaneously injected with B16F10 [PPP/CpG/HA] = 0.75 mg kg−1 I: 808 nm, 1.5 W cm−2, 5 min |
≈20-fold tumor vol.↓ Largest apoptotic cell area CD80+ DC ≈ 3% CD86+ DC ≈ 4.5% |
[107] |
|
Silicon Dioxide |
CuS loaded inside the pores PDMAEMA surface adsorption |
IL-12 gene |
157 nm |
B16F10 cells [CSP] = 34.5 μg mL−1 I: 1064 nm, 0.65 W cm−2, 5 min |
<20% cell viability (CCK-8 assay) 89% apoptotic cells (Annexin V-FITC) ↑CRT expression 66% DCs maturation |
B16F10-bearing C57BL/6 mice [CSP] = 172.4 μg per mouse I: 1064 nm, 0.65 W cm−2, 5 min |
≈3-fold tumor vol.↓ Prolonged survival DC maturation level: ≈48% 21% CD4+ and 12% CD8+ T populations |
[108] |
|
Silicon Dioxide |
Chemical synthesis of UCNP@m-SiO2@liposome NPs |
Ce6 and BSO |
≈50 nm |
B16/F10 cells [UCB] = 100 μg mL−1 I: 980 nm, 0.7 W cm−2, 10 min |
≈29% cell viability (CCK-8 assay) ≈39% apoptosis rate (Western Blot) ↑TNF-α, IL-6, INF-γ |
C57BL/6 mice subcutaneously injected with B16F10 [UCB] = 0.8 mg per mouse I: 980 nm, 0.7 W cm−2, 20 min |
≈3-fold tumor vol.↓ ↑IL-12p40, INF-γ |
[109] |
Abbreviations: ↑, increase; ↓, decrease; aPDL1, anti-programmed death-ligand 1; BSA, bovine serum albumin; B16 cells, B16 murine melanoma cell line; BSO, buthionine sulfoximine B16F10 cells, B16F10 murine melanoma cell line; B16-F10 cells, B16–F10 murine melanoma cell line; B16/F10 cells, B16/F10 murine melanoma cell line; CCK-8, cell counting kit-8; Ce6, chlorin e6; CQ, chloroquine; DC, dendritic cells; HA, hyaluronic acid; IDO, indoleamine 2,3-dioxygenase; M2pep, M2 macrophage-targeting peptide; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide; N/A, not applicable; PDMAEMA, poly((2-di- methylamino)ethyl methacrylate); PEG, polyethylene glycol; PEI, polyethyleneimine; R837, imiquimod; SV, simvastatin; TRP-2, tyrosinase-related protein 2.