Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Conner Chen and Version 1 by Hongna Wang.
Lysosomes are essential organelles of eukaryotic cells and are responsible for various cellular functions, including endocytic degradation, extracellular secretion, and signal transduction. There are dozens of proteins localized to the lysosomal membrane that control the transport of ions and substances across the membrane and are integral to lysosomal function. Mutations or aberrant expression of these proteins trigger a variety of disorders, making them attractive targets for drug development for lysosomal disorder-related diseases.
- lysosome
- drug
- lysosomal membrane protein
1. Introduction to Lysosomes
In 1949, Christian de Duve, a biologist from Brussels, Belgium, set out to investigate how glucose-6-phosphatase responds to insulin regulation by determining its localization in the cell. He isolated various organelles from rat liver homogenates and was surprised to find glucose-6-phosphatase distributed on a new, previously unidentified organelle [8][1]. De Duve, in collaboration with Novikof, observed this organelle with electron microscopy for the first time in 1955 and then named it lysosome. In 1974, De Duve received the Nobel Prize in Physiology or Medicine for discovering lysosomes.
Lysosomes are formed by the fusion of transport vesicles carrying acidic hydrolases budded from the trans-Golgi network with endosomes, which contain molecules taken up by endocytosis at the plasma membrane. Lysosomes are the cell’s reservoir of hydrolases, and their lumen has more than 60 hydrolases that are active only at an acidic pH. Depending on the substrate degraded, lysosomal hydrolases can be classified into several species, including sulfatases, glycosidases, peptidases, phosphatases, lipases, and nucleases, which ensure that lysosomes efficiently degrade a broad range of substances delivered to them, such as mucopolysaccharides, sphingolipids, glycogen, and proteins [9][2]. The membrane-bound vacuolar ATPase (V-ATPase) continuously pumps H+ into the lysosomal lumen by hydrolyzing ATP to maintain its internal pH between 4.5 and 5.5. This acidic environment is critical for the lysosome to maintain its structural integrity and functions of hydrolase activation, calcium storage, vesicle transport, nutrient sensing, and signal transduction. In addition to V-ATPase, lysosomes contain approximately 50 membrane proteins, the most abundant of which are LAMP (lysosomal-associated membrane protein)1, LAMP2, LIMP (lysosomal integral membrane protein)-2, and cluster of differentiation (CD63) [10][3]. The intraluminal portions of these membrane proteins are highly glycosylated, forming a glycocalyx that protects the lysosomal membrane from digestion by acidic intraluminal hydrolases.
2. Lysosomal Degradation
The unique composition and structure of lysosomes allow them to carry out various cellular functions such as degradation, secretion, and signaling. Unlike proteasomes, which degrade only proteins, lysosomes digest multiple substances, including proteins, glycosaminoglycans, nucleic acids, oligosaccharides, and complex lipids. The degradation substrates can be delivered to the lysosome through endocytosis, phagocytosis, or autophagy (Figure 1) [11][4]. Phagocytosis is restricted to specific mammalian cells called phagocytes (usually immune cells), whose function is to remove large pathogens such as bacteria and viruses, dead cell debris, and large dust particles. Endocytosis, which absorbs fluid and solute, on the other hand, occurs in all cells. Endocytosis/phagocytosis absorbs extracellular or surface cargos by plasma membrane-derived endocytic vesicles, which develop over time into early and late endosomes. Mature late endosomes fuse with lysosomes to form a hybrid structure called endolysosome, which carries out the bulk of degradation [12][5].
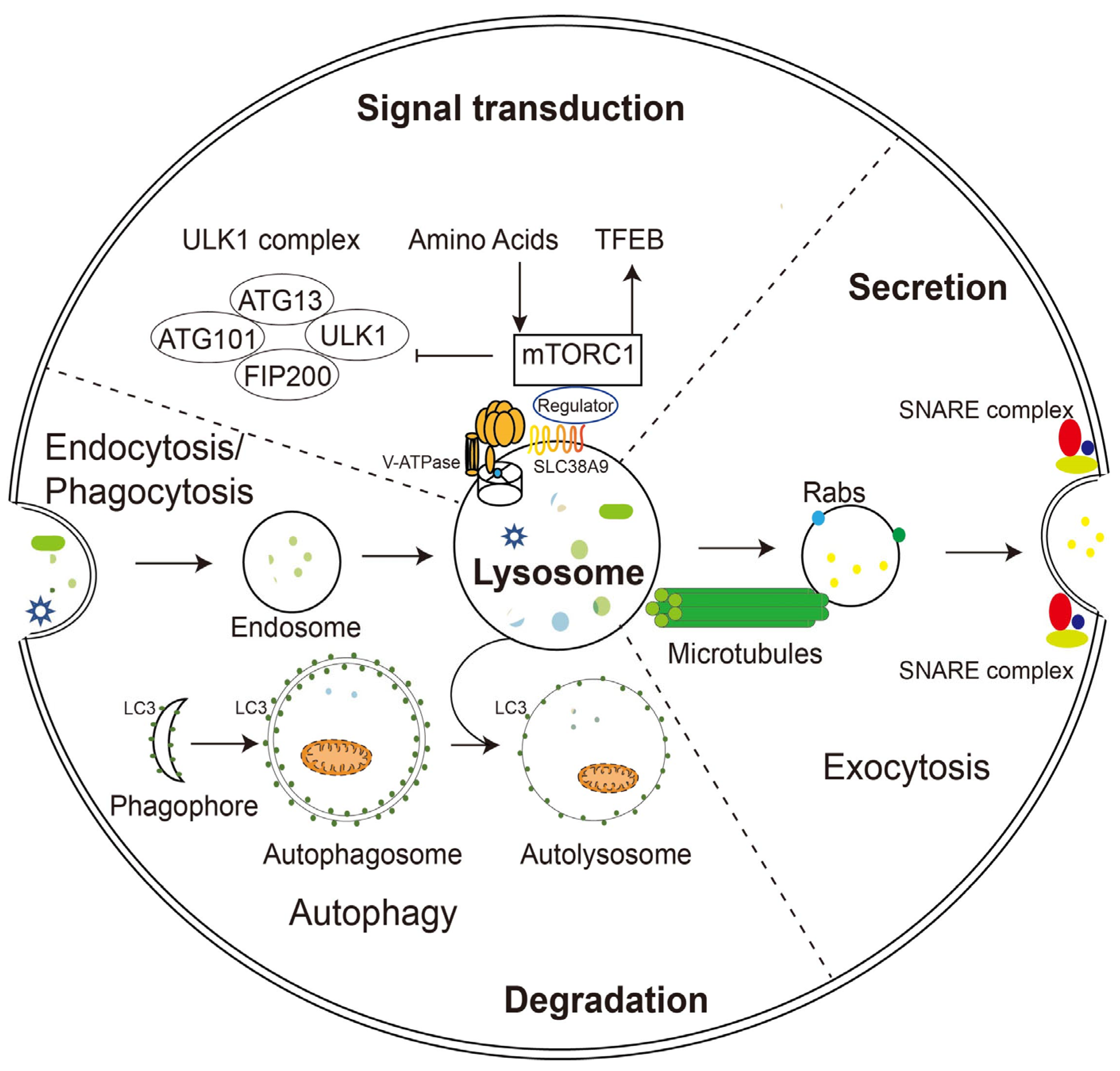
Figure 1. Schematic illustration of lysosomal functions. Lysosomes play a key role in endocytosis/phagocytosis, degradation, exocytosis, and signaling. Nutrients or foreign substances absorbed by endocytosis/phagocytosis enter the cytoplasm through the endosomal/lysosomal pathway. V-ATPase and SLC38A9 on the lysosomal membrane regulate nutrient signaling, TFEB, and autophagy mechanisms by interacting with mTORC1. During phagocytosis, lysosomes move along microtubules and fuse with the plasma membrane with the help of Rabs and SNARE complexes.
In parallel, harmful substances inside the cell, such as aggregated proteins and degenerated organelles, are sent to the lysosome for removal by autophagy (Figure 1). Depending on the mechanism by which degradation substrates are delivered to the lysosome, mammalian autophagy can be divided into three subtypes: in microautophagy, the lysosome engulfs cytoplasmic material through direct membrane invagination; chaperone-mediated autophagy (CMA) uses molecular chaperones carrying cargos to cross the lysosomal membrane directly without membrane remodeling with the assistance of the lysosomal protein LAMP2A; macroautophagy (the most well-studied and often referred to as autophagy) utilizes a particular double-membrane vesicle called autophagosome to encapsulate the cargo, and the autophagosomes eventually fuse with lysosomes to form autolysosomes where the bulk degradation occurs [13,14,15][6][7][8].
3. Lysosomal Exocytosis
In addition to degradation, lysosomes can also extracellularly release their contents in the form of lysosomal exocytosis (Figure 1). During this process, lysosomes migrate along microtubules from the perinuclear region toward the vicinity of the plasma membrane. They then dock and fuse directly with the plasma membrane through mechanisms involving the trans-SNARE complex formation and local Ca2+ release, and offload their contents into the extracellular compartment. Soluble N-ethylmaleimide-sensitive factor attachment protein receptors (SNAREs) are a group of membrane-associated proteins that play a key role in vesicle trafficking and fusion in eukaryotic cells. When a vesicle approaches its target membrane, the SNARE proteins on the vesicle (v-SNAREs) interact with complementary SNARE proteins on the target membrane (t-SNAREs), forming a stable complex that brings the two membranes together. The trans-SNARE complex is formed when v-SNAREs and t-SNAREs interact across two different membranes, leading to membrane fusion and the release of vesicle contents into the target compartment [16][9]. During fusion with the plasma membrane, some lysosomal membrane elements were retained and became a component of the plasma membrane, suggesting the potential of lysosomal exocytosis to remodel the cell membrane [1,17][10][11].
Lysosomal exocytosis is associated with two essential cellular functions, namely membrane repair/remodeling and secretion. Damage to the plasma membrane induces a rapid translocation of lysosomes to the damaged zone. After the local release of Ca2+, lysosomes undergo a series of events that culminate in the fusion of lysosomal membranes with the damaged plasma membrane. This fusion allows for the insertion of lysosomal membrane components into the damaged area, effectively repairing the plasma membrane. At the same time, lysosomal enzymes secreted extracellularly by lysosomes also promote endocytosis of the damaged membrane fragments, thus completing the repair process. Membrane repair occurs in all cell types, while membrane remodeling only occurs in specialized cells. It performs specific functions, for example, the cell membrane extension when macrophages engulf pathogens and the rapid elongation of the apical plasma membrane when neurites outgrow [18][12].
The secretion of lysosomal contents has also been indicated to be involved in different functions, most of which are related to the remodeling of the extracellular matrix by hydrolases released by lysosomal exocytosis. Although there is some evidence that these functions are common to all cell types, early studies overwhelmingly point to them being cell type specific, examples include the degranulation of cytotoxic T-lymphocytes [19][13], bone resorption by osteoclasts [20][14], parasite defense by eosinophils and mast cells [21,22][15][16], melanocyte pigmentation [23][17], platelet coagulation, and the release of sperm hydrolase during fertilization [24][18].
4. Signaling Function of Lysosomes
In addition to its long-recognized function as a recycling station for nutrient production, lysosome has been found to sense intracellular energy and extracellular nutrient status directly and to serve as a signaling hub for coordinating cellular catabolic and anabolic reactions (Figure 1). The lysosomal membrane provides channels (such as V-ATPase and SLC38A9) for nutrients and energy to regulate the mammalian target of rapamycin complex 1 (mTORC1) and the transcriptional factor EB (TFEB), two of the most crucial nutrient-sensitive protein complexes, as well as a dynamic platform for their assembly and activation [3,25][19][20].
mTORC1 is a master regulator of cell growth and metabolism and is activated only in the presence of growth factors and nutrients. When nutrients are adequate, mTOR is recruited to the lysosomal membrane, and kinase activity is initiated. Conversely, when nutrients are lacking, mTOR is inactivated and released from the lysosomal surface. TFEB, one of the best-known downstream effectors of mTORC1, is recruited to the lysosomal membrane and phosphorylated by active mTORC1 under nutrient-rich conditions, thus preventing it from entering the nucleus. When nutrient deficient and mTOR inactivated, TFEB is dephosphorylated and migrates to the nucleus, initiating the transcription of downstream genes that encodes lysosomal hydrolases and lysosomal membrane proteins. The high expression of these genes is fundamental for lysosomal biogenesis and autophagy [26,27][21][22].
TFEB binds directly to a specific E-box-like palindrome sequence called the coordinated lysosomal expression and regulation (CLEAR) motif, found in the promoter regions of most lysosomal genes and many autophagy genes. Therefore, when activated, it coordinates the expression of a wide range of proteins involved in lysosomal biogenesis and function, autophagy, and lysosomal exocytosis. Importantly, TFEB does not regulate the basal transcripts of its targets, but rather enhances their transcript levels in response to environmental cues such as nutrient depletion or stress. Overexpression of TFEB substantially increases the autophagic degradation of substrates such as long-lived proteins, lipid droplets, and damaged mitochondria, suggesting that this transcription factor has regulatory effects on both non-selective and selective autophagy [28][23].
Recent studies have linked the pathogenesis of multiple LSDs and the late-onset of neurodegenerative diseases to impaired autophagy and the accumulation of substrates that fail to degrade. Interestingly, autophagy defects in these pathological processes are often caused by the dysregulation of TFEB. TFEB has been shown to be involved in the clearance of protein aggregates, including β-amyloid and alpha-synuclein, which are hallmark features of neurodegenerative diseases such as Alzheimer’s disease, Parkinson’s disease, and Huntington’s disease. In addition, the overexpression of TFEB ameliorates the severity of disease phenotypes observed in several cellular and mouse models of lysosomal storage diseases (LSDs), including but not limited to polysulfatase deficiency, Batten disease, Pompe disease, Gaucher disease, and cystinosis. This improvement occurs through enhanced lysosomal function and the autophagic clearance of the accumulated substances [29][24]. These studies summarize the role of TFEB as a master regulator of lysosomal biogenesis and autophagy, and its pivotal function in the maintenance of cellular homeostasis and disease pathogenesis.
5. Restoration of Lysosomal Function
As the degradation center of the cell, the lysosome must maintain structural integrity to avoid the devastating consequences of leakage of the dozens of hydrolytic enzymes it houses. Under normal conditions, glycosylation of lysosomal membrane proteins is sufficient to maintain membrane stability against damage by luminal proteolytic enzymes. However, after continuous exposure to oxygen radicals, optical damage, and other irritations, the lysosomal membrane loses its integrity, leading to increased permeability and release of hydrolytic enzymes into the cytoplasm. Without timely repair, sustained lysosomal rupture may lead to the massive release of lysosomal contents, extensive acidification of the cytoplasm and cascade hydrolysis of the contents, and irreversible cellular damage. Early studies showed that moderate lysosomal damage could induce apoptosis, whereas extensive damage led to irreversible necrosis of a large number of cells. Subsequent evidence suggests that sustained lysosomal damage is closely associated with the development of almost all modes of cell death, including apoptosis, necrosis, pyroptosis, and ferroptosis. The specific type of death correlates with the type of cell and the degree of lysosomal damage [30][25].
To maintain their well-being and homeostasis, cells have developed a range of quality control mechanisms for damaged lysosomes, including repair, elimination, and regeneration. The endosomal sorting transport complex (ESCRT), a conserved transport system commonly found in eukaryotic cells, plays a key role in repairing damaged lysosomal membranes. ESCRT relies on its components apoptosis-associated gene-2-interacting protein X (ALIX) and tumor susceptibility gene 101 (TSG101) to tether the lysosomal membrane, and Ca2+ efflux from the ruptured lysosomes enhances the tethering efficiency [31][26]. Exactly how ESCRT repairs lysosomal membranes is unknown, but it may do so by inducing the formation of filamentous helices on the membrane surface and the contraction of the lipid bilayer.
When the lysosomal membrane is damaged beyond repair, it is eliminated by activating a selective form of autophagy called lysophagy. Proteins of the galectin (Gals) family play an important role in lysophagy. Gal3 senses the rupture and binds to the glycoproteins in the lysosomal membrane. After aggregation at the damage site, Gal3 is ubiquitinated and recruits preexisting phagophores for autophagy by binding P62 and LC3. It also recruits autophagy regulatory proteins such as uncoordinated-51-like kinase 1 (ULK1), Beclin 1, and autophagy-related protein 16L1 (ATG16L1) to de novo synthesize phagophore and further amplify lysophagy. Gal8 and Gal9 can also mediate lysophagy, but their mechanism of action is slightly different from that of Gal3 [32,33,34][27][28][29].
With the removal of damaged lysosomes by lysophagy and the reduction in the overall cellular lysosomal population, TFEB is activated to initiate the lysosomal regeneration. The process of lysosome biogenesis has been discussed in the previous section and will not be repeated here.
References
- de Duve, C. The Lysosome Turns Fifty. Nat. Cell Biol. 2005, 7, 847–849.
- Saftig, P.; Haas, A. Turn up the Lysosome. Nat. Cell Biol. 2016, 18, 1025–1027.
- Eskelinen, E.L.; Tanaka, Y.; Saftig, P. At the Acidic Edge: Emerging Functions for Lysosomal Membrane Proteins. Trends Cell Biol. 2003, 13, 137–145.
- Birgisdottir, Å.B.; Johansen, T. Autophagy and Endocytosis—Interconnections and Interdependencies. J. Cell Sci. 2020, 133, jcs228114.
- Saffi, G.T.; Botelho, R.J. Lysosome Fission: Planning for an Exit. Trends Cell Biol. 2019, 29, 635–646.
- Levine, B.; Kroemer, G. Biological Functions of Autophagy Genes: A Disease Perspective. Cell 2019, 176, 11–42.
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of Cells and Tissues. Cell 2011, 147, 728–741.
- Kaushik, S.; Cuervo, A.M. The Coming of Age of Chaperone-Mediated Autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 365–381.
- Jahn, R.; Scheller, R.H. SNAREs—Engines for Membrane Fusion. Nat. Rev. Mol. Cell Biol. 2006, 7, 631–643.
- Trivedi, P.C.; Bartlett, J.J.; Pulinilkunnil, T. Lysosomal Biology and Function: Modern View of Cellular Debris Bin. Cells 2020, 9, 1131.
- Verhage, M.; Toonen, R.F. Regulated Exocytosis: Merging Ideas on Fusing Membranes. Curr. Opin. Cell Biol. 2007, 19, 402–408.
- Pu, J.; Guardia, C.M.; Keren-Kaplan, T.; Bonifacino, J.S. Mechanisms and Functions of Lysosome Positioning. J. Cell Sci. 2016, 129, 4329–4339.
- Stinchcombe, J.C.; Griffiths, G.M. Secretory Mechanisms in Cell-Mediated Cytotoxicity. Annu. Rev. Cell Dev. Biol. 2007, 23, 495–517.
- Väänänen, H.K.; Zhao, H.; Mulari, M.; Halleen, J.M. The Cell Biology of Osteoclast Function. J. Cell Sci. 2000, 113, 377–381.
- Wesolowski, J.; Paumet, F. The Impact of Bacterial Infection on Mast Cell Degranulation. Immunol. Res. 2011, 51, 215–226.
- Logan, M.R.; Odemuyiwa, S.O.; Moqbel, R. Understanding Exocytosis in Immune and Inflammatory Cells: The Molecular Basis of Mediator Secretion. J. Allergy Clin. Immunol. 2003, 111, 923–932.
- Ren, Q.; Ye, S.; Whiteheart, S.W. The Platelet Release Reaction: Just When You Thought Platelet Secretion Was Simple. Curr. Opin. Hematol. 2008, 15, 537–541.
- Tulsiani, D.R.P.; Abou-Haila, A.; Loeser, C.R.; Pereira, B.M.J. The Biological and Functional Significance of the Sperm Acrosome and Acrosomal Enzymes in Mammalian Fertilization. Exp. Cell Res. 1998, 240, 151–164.
- Lamming, D.W.; Bar-Peled, L. Lysosome: The Metabolic Signaling Hub. Traffic 2019, 20, 27–38.
- Napolitano, G.; Di Malta, C.; Ballabio, A. Non-Canonical MTORC1 Signaling at the Lysosome. Trends Cell Biol. 2022, 32, 920–931.
- Condon, K.J.; Sabatini, D.M. Nutrient Regulation of MTORC1 at a Glance. J. Cell Sci. 2019, 132, jcs222570.
- Wolfson, R.L.; Sabatini, D.M. The Dawn of the Age of Amino Acid Sensors for the MTORC1 Pathway. Cell Metab. 2017, 26, 301–309.
- Napolitano, G.; Ballabio, A. TFEB at a Glance. J. Cell Sci. 2016, 129, 2475–2481.
- Bajaj, L.; Lotfi, P.; Pal, R.; Ronza, A.D.; Sharma, J.; Sardiello, M. Lysosome Biogenesis in Health and Disease. J. Neurochem. 2019, 148, 573–589.
- Zhu, S.Y.; Yao, R.Q.; Li, Y.X.; Zhao, P.Y.; Ren, C.; Du, X.H.; Yao, Y.M. Lysosomal Quality Control of Cell Fate: A Novel Therapeutic Target for Human Diseases. Cell Death Dis. 2020, 11, 817.
- Vietri, M.; Radulovic, M.; Stenmark, H. The Many Functions of ESCRTs. Nat. Rev. Mol. Cell Biol. 2020, 21, 25–42.
- Yang, H.; Tan, J.X. Lysosomal Quality Control: Molecular Mechanisms and Therapeutic Implications. Trends Cell Biol. 2023, 1–16.
- Johannes, L.; Jacob, R.; Leffler, H. Galectins at a Glance. J. Cell Sci. 2018, 131, jcs208884.
- Jia, J.; Claude-Taupin, A.; Gu, Y.; Choi, S.W.; Peters, R.; Bissa, B.; Mudd, M.H.; Allers, L.; Pallikkuth, S.; Lidke, K.A.; et al. Galectin-3 Coordinates a Cellular System for Lysosomal Repair and Removal. Dev. Cell 2020, 52, 69–87.e8.
More