Fibroblast growth factors (FGFs) encode a large family of growth factor proteins that activate several intracellular signaling pathways to control diverse physiological functions. The human genome encodes 22 FGFs that share a high sequence and structural homology with those of other vertebrates. FGFs orchestrate diverse biological functions by regulating cellular differentiation, proliferation, and migration. Dysregulated FGF signaling may contribute to several pathological conditions, including cancer. Notably, FGFs exhibit wide functional diversity among different vertebrates spatiotemporally. A comparative study of FGF receptor ligands and their diverse roles in vertebrates ranging from embryonic development to pathological conditions may expand our understanding of FGF.
- fibroblast growth factors
- diversity
- diseases
1. Introduction
2. Gene Organization and Protein Length
Most Fgf genes are dispersed throughout the vertebrate genome. The human and mouse Fgf families comprise 22 members, and the Xenopus Fgf family codes for approximately 19–20 FGFs [7,12,13][7][12][13]. The location of Human Fgf15, mouse Fgf19, Xenopus Fgf15, Fgf17, Fgf18, and Fgf21 has not yet been identified. Moreover, evolutionarily, Fgf15 and Fgf19 are orthologs in vertebrates; human Fgf19 and mouse Fgf15 share 51% amino acid identity, and Xenopus Fgf19 and mouse Fgf15 share 59% identity. Additionally, a few Fgf genes are clustered in the vertebrate genome, including Fgf3, Fgf4, and Fgf19 (Fgf15 in mice), grouped on chromosome 11 in humans and chromosome 7 in mice. However, these clustered associations of Fgfs are common in lower vertebrates, such as Xenopus; Fgf3, and Fgf4, and Fgf19 are closely linked on chromosome 4, and Fgf1, Fgf6, Fgf7, and Fgf23 are grouped on chromosome 3. Notably, Fgf3, Fgf4, and Fgf19 are separated by 30 kb and 45 kb on chromosome 4 in Xenopus; however, this distance reduces to 40 kb and 10 kb in human chromosome 11. In humans and Xenopus, these gene locations indicate a conserved evolutionary pattern conferred by gene and chromosomal duplication and gene translocation. Prototypic Fgfs consist of three coding regions (exons), and this number is relatively conserved in humans, mice, and Xenopus. Exon 1 mainly contains the start codon (ATG); however, there are few Fgfs (Fgf2 and Fgf3) where the sequence initiates from an additional 5′-transcribed sequence upstream of ATG [14,15][14][15]. Additionally, sub-exons are formed in some Fgfs during the splicing process of Exon 1. The gene size of Fgfs varies from <2 kb (in Fgf21) to over 500 kb (in Fgf14). Moreover, unlike other Fgf genes, the Fgf8 exon 1 is subdivided into four small exons in mammals [16] followed by typical exons 2 and 3, reflecting the multifunctionality of the Fgf8 gene. Comparing the genomic sequence of Fgf8 genes from various species reveals that the last three exons are substantially conserved despite the upstream exons being very diverse [16]. Based on the phylogeny chromosomal location (synteny) and homology, the Fgf gene family in humans, mice, and Xenopus can be categorized into seven subfamilies [17], including Fgf1, Fgf4, Fgf7, Fgf8, Fgf9, Fgf11, and Fgf19/15 (Figure 1). Phylogenetic studies suggest potential evolutionary and transformative relationships within the vertebrate gene family. Moreover, studying gene loci on chromosomes allows the evaluation of more precise evolutionary relationships within the Fgf gene family. Lastly, the protein length of FGF is in the range of 126–268 amino acids (aa) in vertebrates, and FGFs in vertebrates are mostly of similar size; therefore, they are predicted to be similarly structured.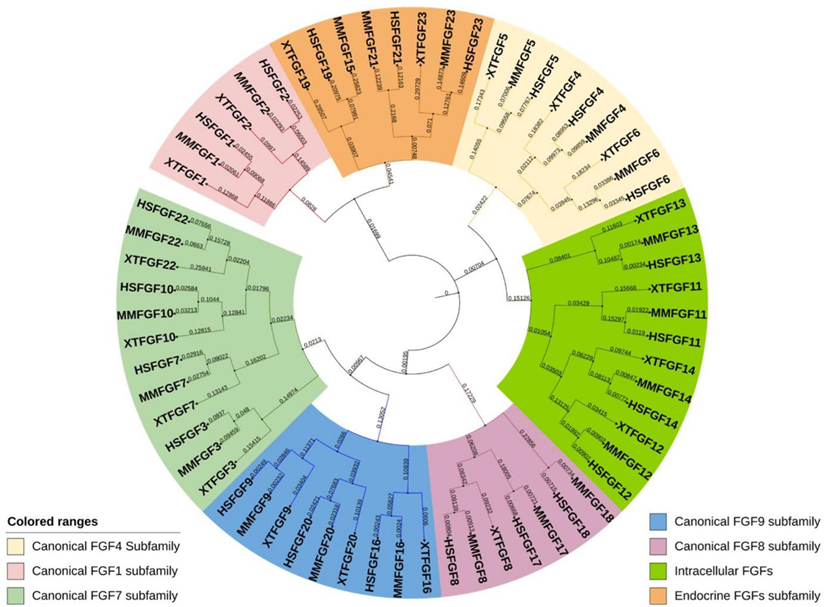
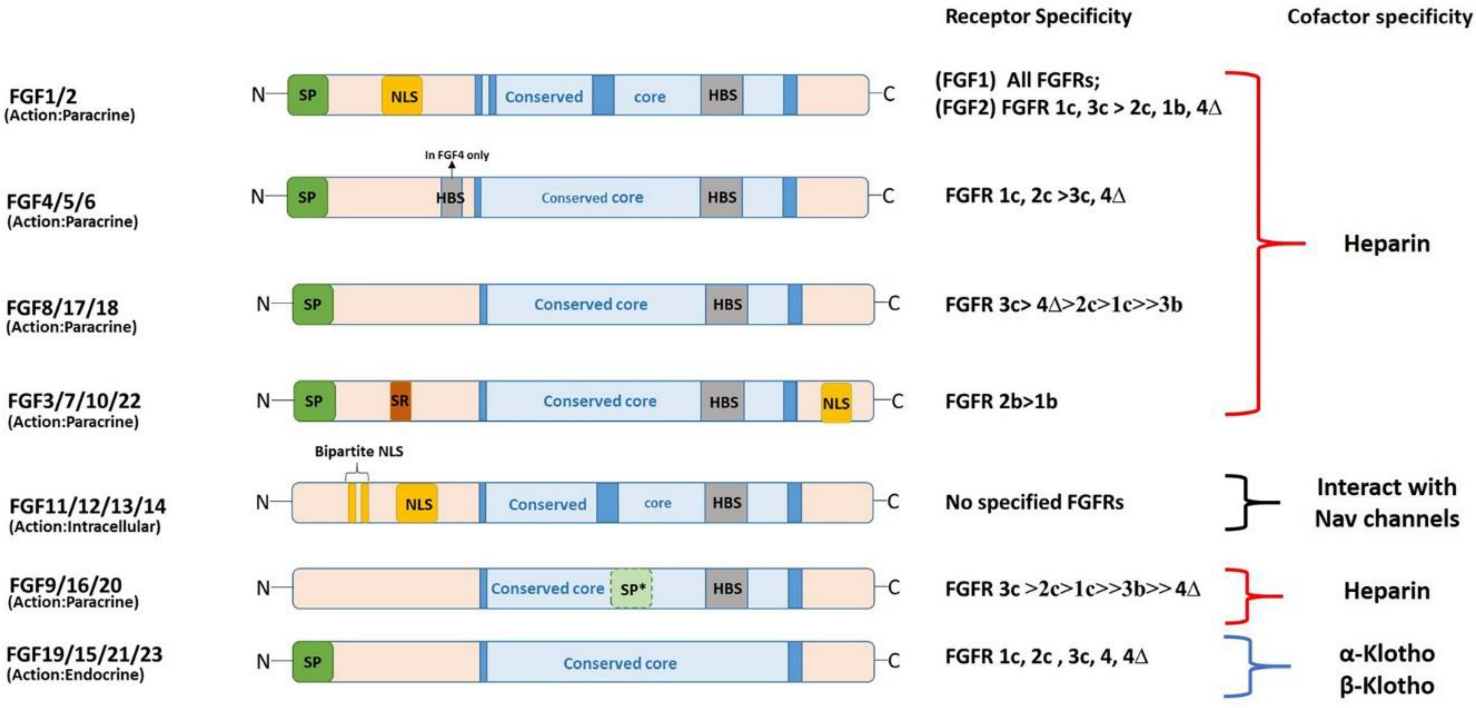
3. Structural and Functional Diversity
4. FGF Signaling in Early Development
4.1. FGF and Mesoderm Specification
Earlier investigations in the 1990s on Xenopus and other vertebrate models showed that FGF signaling is necessary for the formation of the axial (which later forms the notochord) and paraxial mesoderms (which develops into the axial skeleton, muscles, and dermis) [60,64][34][35]. Inhibiting FGF signaling by expressing a dominant negative form of the FGF receptor (Dn-FGFR) disrupts the notochord and somites [60,64,65][34][35][36]. It is unclear whether FGF functions during the induction of axial and paraxial mesoderm or it is required for the maintenance of these mesodermal subtypes. Fletcher and Harland [65][36] reported this dilemma in 2008, when they showed in their investigation that the induction of the paraxial mesoderm requires FGF, and axial mesoderm only requires FGF for maintenance during gastrulation. The FGF requirement for notochord development is evolutionarily conserved in vertebrates [66][37].4.2. FGF and Neural Specification
The spinal cord cells in vertebrates are derived from neuromesodermal progenitors (NMP) with neural and mesodermal features [73,74][38][39]. Events of spinal cord development constitute complex processes, such as neurogenesis, ventral patterning, neural crest specification, and migration, governed by the elongation of the caudal axis [75][40]. Additionally, spinal cord specification involves the FGF signaling pathway as a key regulator. During chicken spinal cord specification, FGF3, FGF4, FGF8, FGF13, and FGF18 are expressed in the caudal NMP region and tissues surrounding the NMPs [76,77][41][42]. FGF8 and FGF4 expression in the NMP region is sustained for several days, and then declines during the last stage of somitogenesis and the cessation of axis elongation [75][40]. FGF/Ras/Mapk/Ets initiate neural induction in ascidians, which are the last common ancestor of vertebrates in chordate evolution [72,81][43][44]. Studies in Xenopus embryos have set the foundation for the classical model (default model) of neural induction, which suggests that signals from the organizer instruct the ectoderm towards neural fate [82][45]. However multiple investigations in chick embryos have established that FGF signaling is vital in early neural differentiation, challenging the default model idea [83,84][46][47]. FGF signaling in neuronal specification can be projected in two ways: first, as an instructive signaling where FGF activates neural genes; second, as antagonist signaling where FGF inhibits BMP signaling via smad1 phosphorylation [12].4.3. FGF Signaling in Metabolism and Diseases (Cancer)
FGF signaling plays a part in the development of almost every organ (including the heart, lungs, brain, urinary system, muscle, skeleton, and skin) and processes such as angiogenesis and lymphangiogenesis [6]. Moreover, endocrine FGFs are functionally essential for metabolism and regulate the brain, kidney, liver, and adipose tissues. The dysregulation of FGF signaling leads to various genetic disorders, including cancer, chronic obstructive pulmonary disease, and chronic kidney disease.4.3.1. FGF Signaling in Metabolism
FGF15/19, FGF21, and FGF23, which belong to the FGF19 subfamily, are endocrine hormones that regulate bile acid, fatty acid, glucose, and mineral metabolisms. Moreover, FGF19 in humans and its ortholog FGF15 are gut-derived circulating hormones that suppress hepatic bile acid via FGFR4 and the cofactor KLB complex [6]. Additionally, FGF15/19 negatively regulates bile acid synthesis and FGF15 deletion in mice upregulates bile acid synthesis by inducing the expression of the rate-limiting and regulating enzyme cholesterol 7α-hydroxylase (CYP7A1) in the liver [104][48]. However, FGF15 overexpression restricts bile acid synthesis by downregulating CYP7A1 mRNA levels [104][48]. FGF15/19 suppresses liver fat storage; in one study, FGF19 transgenic mice showed low levels of lipogenic enzymes and liver triglycerides [107][49]. Moreover, FGF19 blocks lipogenic enzyme expression in rat hepatocytes by inducing STAT3 signaling and suppressing peroxisome proliferator-activated receptor-γ coactivator-1β expression [108][50]. FGF21 is a hormone that regulates glucose and lipid homeostasis and insulin sensitivity. FGF21 functions by binding to FGFR1c and its co-receptor protein KLB in the liver, brain, and adipose tissues [116][51]. FGF21 overexpression in mice resists diet-induced obesity [117][52], and FGF21 can affect weight loss, reduce plasma glucose and triglyceride levels, and boost insulin sensitivity in obese and diabetic vertebrate models without altering the calorie intake [117,118][52][53]. The subcutaneous administration of the FGF21 variant (LY2405319) in DIO mice decreased plasma glucose and body weight at a potency comparable to that of FGF21 [119][54]. FGF23 is a regulator of phosphate metabolism and is produced mainly by the osteoblasts and osteocytes of bone tissue [121][55]. Additionally, FGF23 regulates phosphate and vitamin D homeostasis in skeletal tissues [122][56], and its mutations lead to low serum phosphorus levels, rickets, bone pain, osteomalacia, and short stature [123][57]. Moreover, FGF23 overexpression in whole mouse, and mouse liver and osteoblasts, results in a low serum phosphate concentration and rachitic bone [124,125,126][58][59][60].4.3.2. FGF Signaling in Various Types of Cancer
FGFs are associated with the initiation and progression of cancers, such as multiple myeloma, urothelial carcinoma, hepatocellular carcinoma, and prostate cancer. The FGF1 expression level in several cancer types, such as breast cancer, hepatocellular carcinoma, and esophageal cancer, shows that growth factors promote tumor cell invasion and metastasis [139,140,141][61][62][63]. FGF2 can promote the development of breast cancer cells through ligand-independent activation and the recruitment of estrogen receptor α and PRB4δ4 isoform to MYC regulatory regions [143][64]. Additionally, lung cancer cells that depend on the FGF2/FGFR pathway may be prevented from proliferating using the FGF2 aptamer, which inhibits FGF2 activity [144][65]. In human melanoma produced as a subcutaneous tumor model in nude mice, introducing an episomal vector encoding antisense FGF2 or FGFR1 cDNA could entirely prevent the formation of tumors by blocking angiogenesis [145][66]. Targeting FGF2 to limit melanoma angiogenesis results in decisive anti-melanoma effects, which could lead to novel therapeutic approaches for patients with advanced stages of the disease. FGF4 is expressed more frequently in germ cell cancers, particularly non-seminomas, and may target all-trans-retinoic acid to encourage the growth of malignant-cultured embryonal carcinomas [146][67]. Moreover, increased FGF4 expression is linked to ovarian cancer stem-like cells’ or cancer-initiating cells’ increased capacity to initiate tumors [147][68]. Furthermore, FGF5 is highly expressed in patients with breast cancer [148][69], and FGF6 expression is significantly induced in metastatic liver carcinoma tissues and reduced in non-metastatic liver cancer lesion tissues [149][70].References
- Itoh, N.; Ornitz, D. Functional evolutionary history of the mouseFgf gene family. Dev. Dyn. 2008, 237, 18–27.
- Johnson, D.E.; Williams, L.T. Structural and Functional Diversity in the FGf Receptor Multigene Family. Adv. Cancer Res. 1993, 60, 1–41.
- Morrison, R.S.; Sharma, A.; de Vellis, J.; Bradshaw, R.A. Basic fibroblast growth factor supports the survival of cerebral cortical neurons in primary culture. Proc. Natl. Acad. Sci. USA 1986, 83, 7537–7541.
- Folkman, J.; Klagsbrun, M. Angiogenic factors. Science 1987, 235, 442–447.
- Teven, C.M.; Farina, E.M.; Rivas, J.; Reid, R.R. Fibroblast growth factor (FGF) signaling in development and skeletal diseases. Genes Dis. 2014, 1, 199–213.
- Xie, Y.; Su, N.; Yang, J.; Tan, Q.; Huang, S.; Jin, M.; Ni, Z.; Zhang, B.; Zhang, D.; Luo, F.; et al. FGF/FGFR signaling in health and disease. Signal Transduct. Target. Ther. 2020, 5, 181.
- Ornitz, D.M.; Itoh, N. Fibroblast growth factors. Genome Biol. 2001, 2, REVIEWS3005.
- Ornitz, D.M. FGFs, heparan sulfate and FGFRs: Complex interactions essential for development. Bioessays 2000, 22, 108–112.
- Kuro-O, M. Endocrine FGFs and Klothos: Emerging concepts. Trends Endocrinol. Metab. 2008, 19, 239–245.
- Ornitz, D.M.; Itoh, N. The Fibroblast Growth Factor signaling pathway. Wiley Interdiscip. Rev. Dev. Biol. 2015, 4, 215–266.
- Goetz, R.; Mohammadi, M. Exploring mechanisms of FGF signalling through the lens of structural biology. Nat. Rev. Mol. Cell Biol. 2013, 14, 166–180.
- Kumar, V.; Goutam, R.S.; Park, S.; Lee, U.; Kim, J. Functional Roles of FGF Signaling in Early Development of Vertebrate Embryos. Cells 2021, 10, 2148.
- Itoh, N.; Ornitz, D.M. Evolution of the Fgf and Fgfr gene families. Trends Genet. 2004, 20, 563–569.
- Kiefer, P.; Acland, P.; Pappin, D.; Peters, G.; Dickson, C. Competition between nuclear localization and secretory signals determines the subcellular fate of a single CUG-initiated form of FGF3. EMBO J. 1994, 13, 4126–4136.
- Arnaud, E.; Touriol, C.; Boutonnet, C.; Gensac, M.-C.; Vagner, S.; Prats, H.; Prats, A.-C. A New 34-Kilodalton Isoform of Human Fibroblast Growth Factor 2 Is Cap Dependently Synthesized by Using a Non-AUG Start Codon and Behaves as a Survival Factor. Mol. Cell. Biol. 1999, 19, 505–514.
- Sunmonu, N.A.; Li, K.; Li, J.Y. Numerous isoforms of Fgf8 reflect its multiple roles in the developing brain. J. Cell. Physiol. 2011, 226, 1722–1726.
- Itoh, N.; Ornitz, D.M. Fibroblast growth factors: From molecular evolution to roles in development, metabolism and disease. J. Biochem. 2011, 149, 121–130.
- Powers, C.J.; McLeskey, S.W.; Wellstein, A. Fibroblast growth factors, their receptors and signaling. Endocr.-Relat. Cancer 2000, 7, 165–197.
- Plotnikov, A.N.; Hubbard, S.R.; Schlessinger, J.; Mohammadi, M. Crystal Structures of Two FGF-FGFR Complexes Reveal the Determinants of Ligand-Receptor Specificity. Cell 2000, 101, 413–424.
- Mohammadi, M.; Olsen, S.K.; Ibrahimi, O.A. Structural basis for fibroblast growth factor receptor activation. Cytokine Growth Factor Rev. 2005, 16, 107–137.
- Laestander, C.; Engström, W. Role of fibroblast growth factors in elicitation of cell responses. Cell Prolif. 2014, 47, 3–11.
- Kolli, V.; Paul, S.; Sarkar, N. An Overview on Fibroblast Growth Factors: Structural, Functional and Therapeutic Implications. Curr. Proteom. 2015, 12, 144–151.
- Lea, R.; Papalopulu, N.; Amaya, E.; Dorey, K. Temporal and spatial expression of FGF ligands and receptors during Xenopus development. Dev. Dyn. 2009, 238, 1467–1479.
- Yayon, A.; Klagsbrun, M.; Esko, J.D.; Leder, P.; Ornitz, D.M. Cell surface, heparin-like molecules are required for binding of basic fibroblast growth factor to its high affinity receptor. Cell 1991, 64, 841–848.
- Mohammadi, M.; Olsen, S.K.; Goetz, R. A protein canyon in the FGF–FGF receptor dimer selects from an à la carte menu of heparan sulfate motifs. Curr. Opin. Struct. Biol. 2005, 15, 506–516.
- Sacco, A.; Federico, C.; Giacomini, A.; Caprio, C.; Maccarinelli, F.; Todoerti, K.; Favasuli, V.; Anastasia, A.; Motta, M.; Russo, D.; et al. Halting the FGF/FGFR axis leads to antitumor activity in Waldenström macroglobulinemia by silencing MYD88. Blood 2021, 137, 2495–2508.
- Planque, N. Nuclear trafficking of secreted factors and cell-surface receptors: New pathways to regulate cell proliferation and differentiation, and involvement in cancers. Cell Commun. Signal. 2006, 4, 7.
- Olsnes, S.; Klingenberg, O.; Wie˛dłocha, A. Transport of Exogenous Growth Factors and Cytokines to the Cytosol and to the Nucleus. Physiol. Rev. 2003, 83, 163–182.
- Rodriguez-Enfedaque, A.; Bouleau, S.; Laurent, M.; Courtois, Y.; Mignotte, B.; Vayssière, J.-L.; Renaud, F. FGF1 nuclear translocation is required for both its neurotrophic activity and its p53-dependent apoptosis protection. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2009, 1793, 1719–1727.
- Bouleau, S.; Grimal, H.; Rincheval, V.; Godefroy, N.; Mignotte, B.; Vayssière, J.-L.; Renaud, F. FGF1 inhibits p53-dependent apoptosis and cell cycle arrest via an intracrine pathway. Oncogene 2005, 24, 7839–7849.
- Oulion, S.; Bertrand, S.; Escriva, H. Evolution of the FGF Gene Family. Int. J. Evol. Biol. 2012, 2012, 298147.
- Zhang, X.; Ibrahimi, O.A.; Olsen, S.; Umemori, H.; Mohammadi, M.; Ornitz, D.M. Receptor Specificity of the Fibroblast Growth Factor Family. J. Biol. Chem. 2006, 281, 15694–15700.
- Ornitz, D.M.; Xu, J.; Colvin, J.S.; McEwen, D.G.; MacArthur, C.A.; Coulier, F.; Gao, G.; Goldfarb, M. Receptor Specificity of the Fibroblast Growth Factor Family. J. Biol. Chem. 1996, 271, 15292–15297.
- Amaya, E.; Musci, T.J.; Kirschner, M.W. Expression of a dominant negative mutant of the FGF receptor disrupts mesoderm formation in xenopus embryos. Cell 1991, 66, 257–270.
- Amaya, E.; A Stein, P.; Musci, T.J.; Kirschner, M.W. FGF signalling in the early specification of mesoderm in Xenopus. Development 1993, 118, 477–487.
- Fletcher, R.B.; Harland, R.M. The role of FGF signaling in the establishment and maintenance of mesodermal gene expression in Xenopus. Dev. Dyn. 2008, 237, 1243–1254.
- Fan, T.-P.; Ting, H.-C.; Yu, J.-K.; Su, Y.-H. Reiterative use of FGF signaling in mesoderm development during embryogenesis and metamorphosis in the hemichordate Ptychodera flava. BMC Evol. Biol. 2018, 18, 120.
- Henrique, D.; Abranches, E.; Verrier, L.; Storey, K.G. Neuromesodermal progenitors and the making of the spinal cord. Development 2015, 142, 2864–2875.
- Wilson, V.I. Olivera-Martinez, and K.G. Storey, Stem cells, signals and vertebrate body axis extension. Development 2009, 136, 1591–1604.
- del Corral, R.D.; Morales, A.V. The Multiple Roles of FGF Signaling in the Developing Spinal Cord. Front. Cell Dev. Biol. 2017, 5, 58.
- Delfino-Machín, M.; Lunn, J.S.; Breitkreuz, D.N.; Akai, J.; Storey, K. Specification and maintenance of the spinal cord stem zone. Development 2005, 132, 4273–4283.
- Karabagli, H.; Karabagli, P.; Ladher, R.K.; Schoenwolf, G.C. Comparison of the expression patterns of several fibroblast growth factors during chick gastrulation and neurulation. Anat. Embryol. 2002, 205, 365–370.
- Bertrand, V.; Hudson, C.; Caillol, D.; Popovici, C.; Lemaire, P. Neural Tissue in Ascidian Embryos Is Induced by FGF9/16/20, Acting via a Combination of Maternal GATA and Ets Transcription Factors. Cell 2003, 115, 615–627.
- Miya, T.; Nishida, H. An Ets transcription factor, HrEts, is target of FGF signaling and involved in induction of notochord, mesenchyme, and brain in ascidian embryos. Dev. Biol. 2003, 261, 25–38.
- Spemann, H.; Mangold, H. über Induktion von Embryonalanlagen durch Implantation artfremder Organisatoren. Dev. Genes Evol. 1924, 100, 599–638.
- Wilson, S.I.; Edlund, T. Neural induction: Toward a unifying mechanism. Nat. Neurosci. 2001, 4, 1161–1168.
- Stern, C.D. Neural induction: Old problem, new findings, yet more questions. Development 2005, 132, 2007–2021.
- Kliewer, S.A.; Mangelsdorf, D.J. Bile Acids as Hormones: The FXR-FGF15/19 Pathway. Dig. Dis. 2015, 33, 327–331.
- Tomlinson, E.; Fu, L.; John, L.; Hultgren, B.; Huang, X.; Renz, M.; Stephan, J.P.; Tsai, S.P.; Powell-Braxton, L.; French, D.; et al. Transgenic mice expressing human fibroblast growth factor-19 display increased metabolic rate and decreased adiposity. Endocrinology 2002, 143, 1741–1747.
- Bhatnagar, S.; Damron, H.A.; Hillgartner, F. Fibroblast Growth Factor-19, a Novel Factor That Inhibits Hepatic Fatty Acid Synthesis. J. Biol. Chem. 2009, 284, 10023–10033.
- Tacer, K.F.; Bookout, A.L.; Ding, X.; Kurosu, H.; John, G.B.; Wang, L.; Goetz, R.; Mohammadi, M.; Kuro-O, M.; Mangelsdorf, D.J.; et al. Research Resource: Comprehensive Expression Atlas of the Fibroblast Growth Factor System in Adult Mouse. Mol. Endocrinol. 2010, 24, 2050–2064.
- Coskun, T.; Bina, H.A.; Schneider, M.A.; Dunbar, J.D.; Hu, C.C.; Chen, Y.; Moller, D.E.; Kharitonenkov, A. Fibroblast Growth Factor 21 Corrects Obesity in Mice. Endocrinology 2008, 149, 6018–6027.
- Xu, J.; Lloyd, D.J.; Hale, C.; Stanislaus, S.; Chen, M.; Sivits, G.; Vonderfecht, S.; Hecht, R.; Li, Y.-S.; Lindberg, R.A.; et al. Fibroblast Growth Factor 21 Reverses Hepatic Steatosis, Increases Energy Expenditure, and Improves Insulin Sensitivity in Diet-Induced Obese Mice. Diabetes 2009, 58, 250–259.
- Kharitonenkov, A.; Beals, J.M.; Micanovic, R.; Strifler, B.A.; Rathnachalam, R.; Wroblewski, V.J.; Li, S.; Koester, A.; Ford, A.M.; Coskun, T.; et al. Rational Design of a Fibroblast Growth Factor 21-Based Clinical Candidate, LY2405319. PLoS ONE 2013, 8, e58575.
- Shapter, A.E. The overpass syndrome. Can. Med. Assoc. J. 1992, 146, 113.
- Quarles, L.D. Skeletal secretion of FGF-23 regulates phosphate and vitamin D metabolism. Nat. Rev. Endocrinol. 2012, 8, 276–286.
- Consortium, A. Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. Nat. Genet. 2000, 26, 345–348.
- Shimada, T.; Urakawa, I.; Yamazaki, Y.; Hasegawa, H.; Hino, R.; Yoneya, T.; Takeuchi, Y.; Fujita, T.; Fukumoto, S.; Yamashita, T. FGF-23 transgenic mice demonstrate hypophosphatemic rickets with reduced expression of sodium phosphate cotransporter type IIa. Biochem. Biophys. Res. Commun. 2004, 314, 409–414.
- Larsson, T.; Marsell, R.; Schipani, E.; Ohlsson, C.; Ljunggren, O.; Tenenhouse, H.S.; Jppner, H.; Jonsson, K.B. Transgenic mice expressing fibroblast growth factor 23 under the control of the alpha1(I) collagen promoter exhibit growth retardation, osteomalacia, and disturbed phosphate homeostasis. Endocrinology 2004, 145, 3087–3094.
- Bai, X.; Miao, D.; Li, J.; Goltzman, D.; Karaplis, A.C. Transgenic Mice Overexpressing Human Fibroblast Growth Factor 23 (R176Q) Delineate a Putative Role for Parathyroid Hormone in Renal Phosphate Wasting Disorders. Endocrinology 2004, 145, 5269–5279.
- Szlachcic, A.; Sochacka, M.; Czyrek, A.; Opalinski, L.; Krowarsch, D.; Otlewski, J.; Zakrzewska, M. Low Stability of Integrin-Binding Deficient Mutant of FGF1 Restricts Its Biological Activity. Cells 2019, 8, 899.
- Slattery, M.L.; John, E.M.; Stern, M.C.; Herrick, J.; Lundgreen, A.; Giuliano, A.R.; Hines, L.; Baumgartner, K.B.; Torres-Mejia, G.; Wolff, R.K.; et al. Associations with growth factor genes (FGF1, FGF2, PDGFB, FGFR2, NRG2, EGF, ERBB2) with breast cancer risk and survival: The Breast Cancer Health Disparities Study. Breast Cancer Res. Treat. 2013, 140, 587–601.
- Ribatti, D.; Vacca, A.; Rusnati, M.; Presta, M. The discovery of basic fibroblast growth factor/fibroblast growth factor-2 and its role in haematological malignancies. Cytokine Growth Factor Rev. 2007, 18, 327–334.
- Giulianelli, S.; Riggio, M.; Guillardoy, T.; Pérez Piñero, C.; Gorostiaga, M.A.; Sequeira, G.; Pataccini, G.; Abascal, M.F.; Toledo, M.F.; Jacobsen, B.M.; et al. FGF2 induces breast cancer growth through ligand-independent activation and recruitment of ERalpha and PRBDelta4 isoform to MYC regulatory sequences. Int. J. Cancer 2019, 145, 1874–1888.
- Hamamoto, J.; Yasuda, H.; Nonaka, Y.; Fujiwara, M.; Nakamura, Y.; Soejima, K.; Betsuyaku, T. The FGF2 aptamer inhibits the growth of FGF2-FGFR pathway driven lung cancer cells. Biochem. Biophys. Res. Commun. 2018, 503, 1330–1334.
- Wang, Y.; Becker, D. Antisense targeting of basic fibroblast growth factor and dibroblast growth factor receptor-1 in human melanomas blocks intratumoral angiogenesis and tumor growth. Nat. Med. 1997, 3, 887–893.
- Maerz, W.J.; Baselga, J.; Reuter, V.E.; Mellado, B.; Myers, M.L.; Bosl, G.J.; Spinella, M.J.; Dmitrovsky, E. FGF4 dissociates anti-tumorigenic from differentiation signals of retinoic acid in human embryonal carcinomas. Oncogene 1998, 17, 761–767.
- Yasuda, K.; Torigoe, T.; Mariya, T.; Asano, T.; Kuroda, T.; Matsuzaki, J.; Ikeda, K.; Yamauchi, M.; Emori, M.; Asanuma, H.; et al. Fibroblasts induce expression of FGF4 in ovarian cancer stem-like cells/cancer-initiating cells and upregulate their tumor initiation capacity. Lab. Investig. 2014, 94, 1355–1369.
- Huang, Y.; Wang, H.; Yang, Y. Expression of Fibroblast Growth Factor 5 (FGF5) and Its Influence on Survival of Breast Cancer Patients. Experiment 2018, 24, 3524–3530.
- Guo, S.; Jiang, S.; Epperla, N.; Ma, Y.; Maadooliat, M.; Ye, Z.; Olson, B.; Wang, M.; Kitchner, T.; Joyce, J.; et al. A gene-based recessive diplotype exome scan discovers FGF6, a novel hepcidin-regulating iron-metabolism gene. Blood 2019, 133, 1888–1898.