Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Rok Herman and Version 2 by Catherine Yang.
Polycystic ovary syndrome (PCOS) is the most common endocrine and metabolic disorder in women of reproductive age. Its heterogeneous clinical presentation is characterized by hyperandrogenemia, reproductive changes, polycystic ovary morphology, and insulin resistance (IR). The primary pathophysiological process in its multifactorial etiology has not yet been identified. Although commonly proposed as an initial abnormality, IR is most often looked at in isolation, without the proper investigation of other essential steps in insulin metabolism.
- polycystic ovary syndrome
- PCOS
- insulin resistance
- beta cell function
- insulin clearance
1. Introduction
Polycystic ovary syndrome (PCOS) is the most common endocrine/metabolic disorder in women of reproductive age. It affects up to 20% of women worldwide, and its prevalence has been on the rise over the last decade [1]. Its development and clinical presentation are characterized by multiple underlying metabolic, hyperandrogenic, and reproductive abnormalities. Despite the continuous efforts to identify the primary pathophysiological process, the scientific community is still torn between different proposed theories, and the precise and unifying mechanism remains to be identified. Consequently, the treatment, to a large degree, still depends on lifestyle intervention and symptomatic management of individual signs and symptoms. The new era of anti-obesity therapy offers a novel powerful pharmacologic tool to improve metabolic, reproductive, and other clinical outcomes in a subset of patients who are overweight or obese, primarily through weight reduction [2]. However, a better understanding of individual patient phenotypes and the dominant pathophysiological process is necessary to improve the management of all patients.
2. The Role of Insulin Resistance in PCOS
The coexistence of insulin resistance (IR) and PCOS was first described in 1980, when it was demonstrated that hyperandrogenism correlates with hyperinsulinism in women with PCOS [3][7]. A few years later, a small study found that even lean patients with PCOS are insulin resistant [4][8], which paved the way to the hypothesis that IR might be central to PCOS. Despite this finding, the established PCOS diagnostic criteria continued to focus on hyperandrogenism and ovarian morphology and function, without taking into account the clinical variables related to glucose metabolism. Consequently, a significant degree of metabolic heterogeneity is observed between the four phenotypes, based on Rotterdam diagnostic criteria, with IR being the most dominant proposed etiology in the subgroup of phenotype A patients [5][9]. The current estimates are that up to 75% of PCOS women have impaired insulin response, as measured by the hyperinsulinemic-euglycemic clamp method [6][10]. Despite numerous studies investigating the relationship between excess weight, decreased insulin sensitivity, and hyperinsulinemia in women with PCOS, the understanding of this multidirectional and synergistic web of interactions remains unclear [7][11]. An additional degree of uncertainty comes from the frequent use of different surrogate estimates of IR and less convincing results for the degree of IR in lean PCOS patients in comparison to age- and BMI-matched controls [7][8][9][10][11][12][11,12,13,14,15,16]. Similar to type 2 diabetes, in PCOS, the most commonly proposed sequence of events starts with IR as the primary insult, leading to compensatory hyperinsulinemia that temporarily maintains normal glycemia. However, with disease progression, relative or absolute insulin deficiency presents as prediabetes or T2D in the predisposed patients [6][10]. Throughout this process, a considerable period in the disease course is marked by supraphysiologic insulin levels that directly and indirectly disrupt ovarian function, as well as exert other, not yet fully explored, changes [5][9]. Insulin is able to act synergistically with luteinizing hormone as a co-gonadotrophin within ovarian theca cells by enhancing the production of androgens. Moreover, insulin mediates follicular development, promoting the arrest of pre-antral follicle development in the setting of hyperinsulinemia [5][13][14][9,17,18]. The clinical presentation of PCOS patients is influenced by other insulin related systemic effects through modulation of the luteinizing hormone pulse amplitude, stimulation of adrenal androgen secretion, and the suppression of hepatic sex hormone binding globulin production, which increases the amount of free testosterone [5][13][15][16][9,17,19,20]. As with other insulin-resistant conditions, it remains unresolved whether the decreased insulin activity might be due to an intrinsic defect in the insulin signaling pathway, or is instead induced by environmental factors, which further prevent the evaluation of IR in PCOS, as well as the development of causal interventions [17][21]. Still, the improvement in insulin sensitivity remains one of the primary desired treatment outcomes in most interventions, since extensive data reinforce the correlation between improved insulin sensitivity and other beneficial metabolic and reproductive outcomes [18][19][22,23]. Despite this fact, interventions that specifically targeted the insulin signaling pathway and directly enhanced insulin sensitivity have, to date, provided only limited clinical efficacy. In addition, although the initial studies in the 1980s showed increased levels of IR in lean PCOS patients, many conflicting results were later reported, and the described prevalence of IR is generally lower than that in overweight or obese patients [20][24]. In light of those questions and the importance of hyperinsulinemia alone in PCOS development, it is also essential to mention the potential conceptual shift in the understanding of IR proposed in recent years by some authors and research groups [21][22][23][24][25][26][25,26,27,28,29,30]. However, the amount of further data supporting this viewpoint is limited. The complex position of IR in PCOS pathophysiology and clinical presentation requires a comprehensive analysis of the entire insulin metabolism and its function at different stages. In the following sections, the steps leading from deviations in insulin secretion from pancreatic beta cells, to the mechanisms of impaired insulin action at the target cells, to the reduced insulin clearance are explored in detail. Figure 1 presents a summary of a new view of the role of insulin metabolism in PCOS pathogenesis, with hyperinsulinemia proposed as an important factor in PCOS development.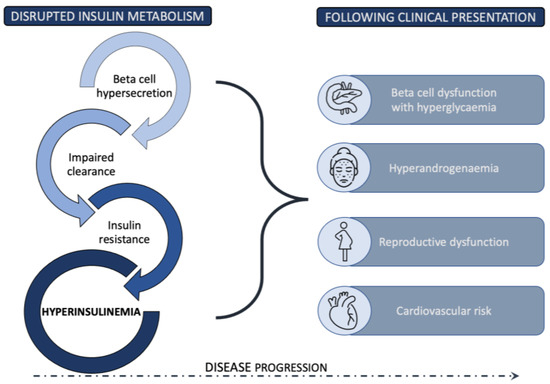
Figure 1.
Disrupted insulin metabolism resulting in hyperinsulinemia as an essential pathophysiological driver in PCOS development.
2. Pancreatic Beta Cell Function and PCOS
Studies have provided controversial results regarding the secretory function of pancreatic beta cells in PCOS patients. Some authors reported defective insulin secretion, whereas others demonstrated increased insulin secretion; however, in general, the data is limited. These discrepancies can, to some extent, be explained by different study protocols, in which research groups reported levels of insulin secretion in either basal or stimulated states, and only some study protocols adjusted the levels of insulin secretion to the prevailing level of IR. An essential aspect when extrapolating insulin levels to the estimates of beta cell function is that the role of insulin clearance also needs to be addressed. In addition, the way in which different stages of PCOS progression are characterized by diverse disturbances in glucose homeostasis is seldom assessed. The occurrence of hyperinsulinemia in PCOS has previously been confirmed by the hyperinsulinemic clamp method. Interestingly, the same method demonstrated the presence of hyperinsulinemia in lean PCOS patients with normal insulin sensitivity, and even higher insulin secretion in lean patients than in obese patients or controls [11][12][15,16]. Furthermore, a comparison between PCOS patients and weight-matched controls demonstrated that PCOS patients had higher basal and cumulative 24 h insulin concentrations, despite having similar glucose concentrations. In contrast, their incremental insulin response to meals was markedly reduced, and further analysis showed that this reduction resulted from a decrease in amplitude, rather than pulse frequency [27][33]. To summarize, the available studies offer conflicting and limited results regarding the beta cell function in PCOS patients and provide little insight into the proper position of this function in PCOS pathogenesis. Heterogeneity in study designs including patients in different stages of the PCOS course; measurements of either basal, stimulated, or cumulative insulin concentrations; and not adjusting for the degree of IR and insulin clearance add to the difficulty of analyzing this topic. However, based on the current data, the role of beta cell function in the development of PCOS and its clinical presentation deserves further investigation.3. Mechanisms of Insulin Resistance
IR is defined by an impaired response to insulin in target tissues—muscle, fat, and liver [28][58]—and predominantly manifests with a decreased utilization of glucose due to a defective glucose transport across the plasma membrane, facilitated by glucose transporter type 4 (GLUT4) [29][59]. IR in PCOS is the result of a post-receptor abnormality due to a disruption in signal transmission downstream from the insulin receptor. Thus, insulin-resistant tissues exhibit decreased responsiveness and sensitivity to insulin stimulation, whereas this effect is more pronounced in PCOS patients than in obese patients [30][31][60,61]. Apart from the metabolic effects of insulin, such as increased glucose uptake, glycogen synthesis, and protein synthesis, which are mediated through the phosphoinositide 3-kinase (PI3K) pathway, it also exhibits mitogenic and steroidogenic effects that are conveyed through the mitogen-activated protein kinase (MAPK) pathway. In patients with PCOS, IR selectively affects only the PI3K pathway, while the MAPK pathway functions normally [32][33][62,63]. Previous studies of IR mechanisms in common insulin-resistant states, such as obesity, type 2 diabetes, and PCOS, have implicated that the pathogenesis of IR in the latter state might be unique [28][30][32][34][58,60,62,64]. In this chapter, thwe researchers only briefly explore the molecular mechanisms associated with IR in PCOS. Since some of the presented findings had been provided by in vivo or in vitro studies on non-human models, translation to the human organism is somewhat limited. In addition, conflicting results from studies in different human body tissues indicate that the insulin signaling and molecular mechanisms behind IR may differ between tissues, restricting generalization. Insulin conveys its effects through the insulin signaling pathway (Figure 2). It starts with insulin binding to the insulin receptor and is followed by the autophosphorylation of the receptor’s tyrosine residues. This leads to the phosphorylation of the insulin receptor substrate (IRS) that conveys the signal downstream through the PI3K pathway and the MAPK pathway. PI3K drives the activation of protein kinase B (PKB or Akt), which interacts with numerous downstream proteins responsible for the metabolic effects of insulin [29][59], including the translocation of GLUT4 [35][65].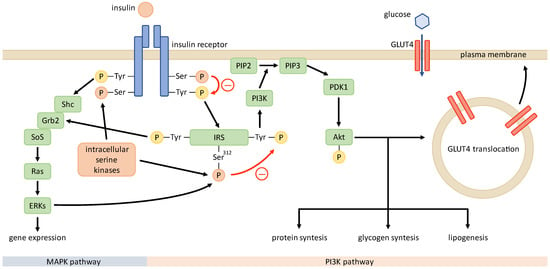
Figure 2. Insulin signaling pathway in PCOS. Unknown intracellular serine kinases may phosphorylate serine residues of insulin receptor and IRS, impairing the phosphorylation of tyrosine residues and signal transduction after stimulation with insulin. This renders the metabolic PI3K pathway defective, while the mitogenic MAPK pathway functions normally.