Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Beatrix Zheng and Version 1 by Giuseppina Martella.
Strong evidence suggests a correlation between degeneration and mitochondrial deficiency. Typical cases of degeneration can be observed in physiological phenomena (i.e., ageing) as well as in neurological neurodegenerative diseases and cancer. All these pathologies have the dyshomeostasis of mitochondrial bioenergy as a common denominator.
- movement disorders
- mitochondria
- energy metabolism
1. Introduction
The mitochondrion is an evolutionary organelle that originated from the established symbiotic relationship between alpha-proteobacteria and eukaryotic cells [1]. This organelle was stabilized in the host through mitotic cell divisions [1,2][1][2]. In light of this hypothesis, it is possible that the mitochondria enhanced the development of life as we know it and represent the common factor through which many organisms function or dysfunction [2,3,4][2][3][4]. Mitochondria are well-known energetic units of eukaryotic cells [2]. They supply energy in the form of ATP to support all cellular functions. In aerobic respiration, through a complex biochemical pathway that involves the addition of different co-factors, the mitochondria aid in the synthesis of ATP molecules from pyruvate [2,3][2][3]. In addition, mitochondrial proteomes, which are aggregates of more than 1000 proteins, can support a wide variety of critical biochemical processes, including amino acid metabolism, nucleotide metabolism, protein synthesis, and fatty acid catabolism [3,4][3][4].
2. Role of Mitochondria in Brain Energy Metabolism, Calcium Homeostasis, and Signal Transduction
Mitochondria are organelles responsible for several critical processes in neuronal function and dysfunction, including energy metabolism, calcium homeostasis, and signal transduction [1]. The brain consumes about 20% of the total oxygen respired to meet the high metabolic demand of neurons that must maintain ionic gradients across membranes, transport molecules from the soma along axons and dendrites, as well as for neurotransmission [12][5]. The metabolic activities that generate ATP rely mainly on mitochondrial oxidative phosphorylation (OXPHOS) [1]. During the process of OXPHOS, enzymes of the Krebs cycle utilize acetyl-coenzyme A to reduce the cofactors, nicotinamide adenine dinucleotides (NADH) and flavin adenine dinucleotides (FADH2), that aid energy transfer to the electron transport chain (ETC), embedded within the extensive inner mitochondrial membrane [13,14][6][7]. The ETC consists of four complexes that transfer electrons from NADH and FADH2 to O2. Ultimately, the energy released by the transfer of electrons is used for the thermodynamically unfavorable pumping of protons against their concentration gradient from the matrix to the intermembrane space to generate an electrochemical gradient known as mitochondrial membrane potential (ΔΨm) [1]. This membrane potential is essential for the process of energy storage (Figure 1). The protons accumulated in the intermembrane space are then allowed to move according to a concentration gradient back into the matrix by passing through one of the domains of the enzyme ATP synthase (or Complex V), which finally harnesses the released energy to phosphorylate ADP molecules into ATP [13][6]. Proper neuronal function is strongly linked to the retention of mitochondrial membrane potential and ATP levels [15][8] (Figure 1).
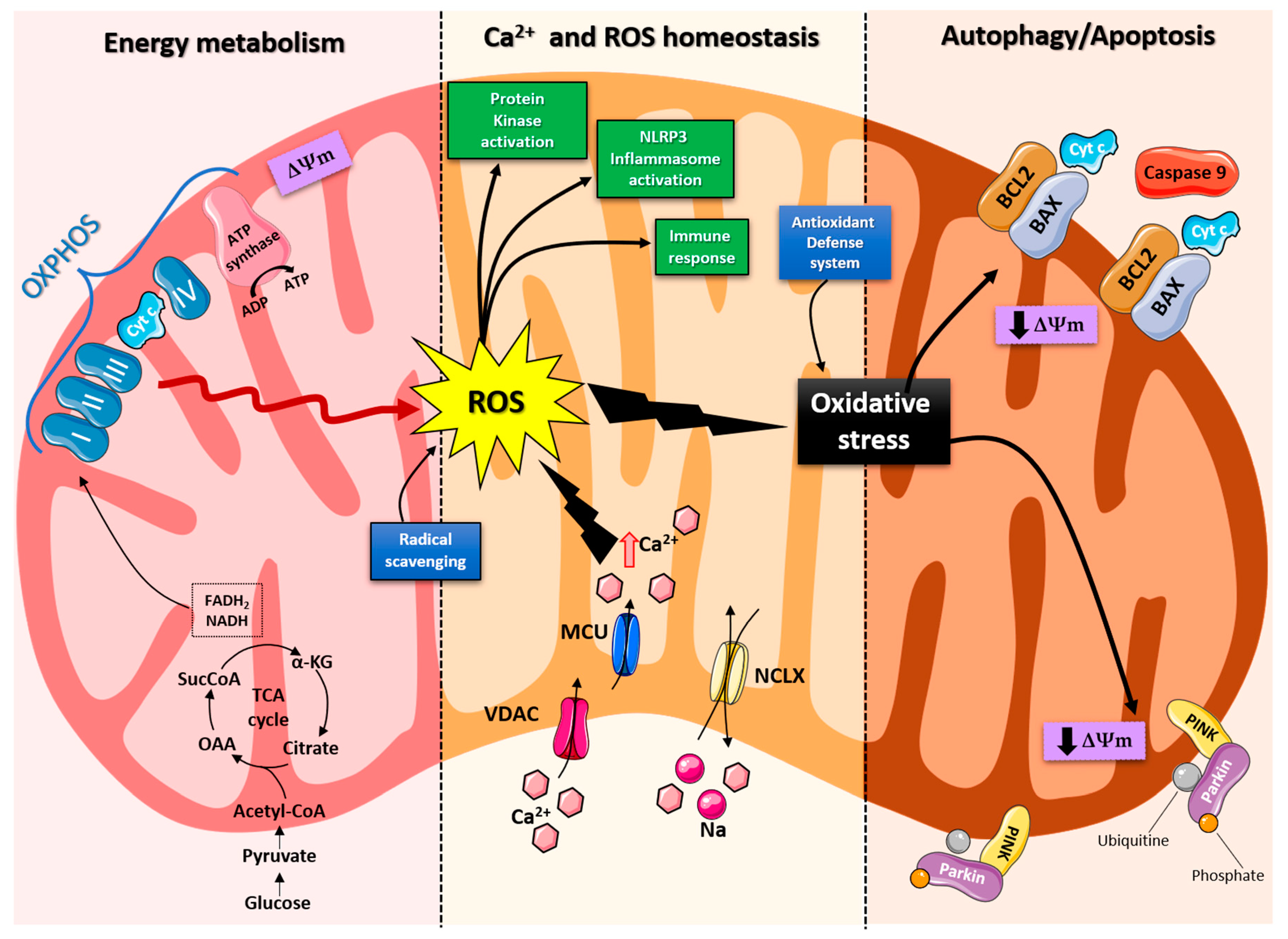
Figure 1. Schematic representation of the mitochondrial functions. From left: Under physiological conditions, mitochondria supply ATP through OXPHOS. Krebs cycle enzymes use acetyl-coenzyme A to reduce NADH and FADH2, which are used for energy transfer to the electron transport chain (ETC) embedded in the inner mitochondrial membrane. OXPHOS is also an important source of ROS, whose basal levels are maintained by the radical scavenging network. Mitochondria also play a crucial role in calcium homeostasis. The voltage-dependent anion channel (VDAC) and the mitochondrial Ca2+ uniporter complex (MCU) finely control Ca2+ passage across the mitochondrial membranes, while the mitochondrial Na+/Ca2+ exchanger (NCLX) is one of the central units involved in Ca2+ extrusion. Under normal conditions, through ROS generation and redox signaling, mitochondria can control cellular metabolism, physiology, the inflammatory response, and immune function and act as important signaling molecules in the cell by activating various protein kinases. In contrast, the overproduction of ROS and dysregulation of the redox signaling system result in oxidative stress that can lead to mitochondrial damage. Malfunctioning mitochondria can be selectively removed through mitophagy, or, as all other defense mechanisms fail, the neuron can orchestrate its own destruction by activating the intrinsic suicide program of apoptosis.
Indeed, mitochondrial functions go beyond the primary role of ATP production because these organelles are intimately involved in numerous processes operating within the cell, including calcium homeostasis, the generation of free radical species (ROS), steroid synthesis, apoptosis, and cell signaling pathways [16][9]. Particularly, the direction of the mitochondrial membrane potential (negative internal) has important implications, as it elicits the thermodynamic force supporting the accumulation of metal cations, especially calcium (Ca2+) in the mitochondria [17][10]. Mitochondrial Ca2+ homeostasis plays a key role in cellular bioenergetics and signaling [18][11]. Ca2+ passage across the outer mitochondrial membrane (OMM) is mediated by the VDAC channel, whose different selectivity for cations or anions is voltage-dependent [19][12]. For instance, at low potentials (10 mV), the VDAC channel is highly permeable to anions and able to maintain a low Ca2+ flux. Conversely, increases in the membrane potential (20–30 mV) result in a conformational change that allows a 4- to 10-fold increase in Ca2+ influx [20][13]. In comparison, the inner mitochondrial membrane (IMM) has considerable Ca2+ permeability: the mitochondrial calcium uniporter (MCU) contributes to the potential-dependent Ca2+ influx into the mitochondrial matrix, while the mitochondrial Na+/Ca2+ exchanger (NCLX) is one of the main units involved in Ca2+ extrusion [21][14]. In general, cytosolic calcium uptake by mitochondria will occur only if they are exposed to elevated Ca2+ concentrations [22,23][15][16] (Figure 1). However, within the cell, Ca2+ is compartmentalized, and the average resting cytosolic concentration is remarkably low [24,25,26][17][18][19]. One of the main stores of cellular calcium is the endoplasmic reticulum (ER), which contributes to orchestrating cellular Ca2+ homeostasis through its interaction with the mitochondria [27][20]. Notably, Ca2+ uptake in the mitochondria probably occurs only at sites in close proximity between the ER and mitochondria [18][11]. On the other hand, neuronal mitochondria, unlike those in non-neuronal cells, have a considerably lower threshold for Ca2+ uptake. In addition, ER-mitochondria contacts are critical for Ca2+ uptake in dendritic mitochondria but not in axonal mitochondria [28][21]. This property of axonal mitochondria is due to the presence of the brain-specific uniporter MICU3, which causes axons to be less dependent on intracellular Ca2+ storage. In the absence of MICU3, synaptic function is impaired [29][22] (Figure 1).
Mitochondrial calcium-signaling in neurons regulates metabolism and energy production, which are crucial for neurotransmission and sustaining synaptic plasticity [30][23]. In addition, mitochondrial Ca2+ also drives the production of reactive oxygen species (ROS) [31][24]. Mitochondria are the primary sources of ROS in cells and actively participate in cellular redox regulation and ROS signaling [32][25] (Figure 1). ROS, in the form of superoxide, are natural byproducts of normal mitochondrial activity and are naturally converted to H2O2, which is in turn scavenged by the enzyme catalase to produce water [32][25]. Under normal conditions, through the generation of ROS and redox signaling, mitochondria can control cell metabolism and physiology, as well as inflammatory responses, immune function, autophagy, and stress responses [33,34,35][26][27][28]. Indeed, ROS, and particularly hydrogen peroxide at low concentrations, act as important signaling molecules in the cell, activating several protein kinases, such as PKA, PKC, PI3K, and p38 [36][29]. Moreover, in immune cells, mitochondrial metabolites and ROS finely regulate signaling pathways and cell fate, thereby orchestrating the immune response [37][30].
The immune system comprises a diverse family of cells with multiple roles during homeostasis and inflammation, capable of using distinct metabolic programs to undertake their functions. For instance, during the immune response, effector T cells promote aerobic glycolysis, while memory T cells and regulatory T cells promote fatty acid oxidation [38][31]. Particularly, the mitochondria can modulate the metabolic and physiological states of different types of immune cells [39][32]. It can also stimulate the innate immune signaling cascade, which can intensify inflammation following cytotoxic stimuli or microbial infection [40][33]. For example, the innate immune receptor NLRX1, a member of the Nod-like Receptor (NLR) family, is located in the mitochondria and undertakes an important role in maintaining cellular homeostasis following acute mitochondrial injury [41,42][34][35]. In addition, it has recently been discovered that mitochondria play a central role in initiating and regulating the NLRP3 (nucleotide-binding domain, leucine-rich repeat family, pyrin domain-containing 3) inflammasome [43][36]. This multiprotein complex, activated upon infection or cellular stress, leads to the secretion of proinflammatory cytokines, such as interleukin-1β (IL-1β) and IL-18, that trigger an inflammatory form of cell death called pyroptosis [44,45][37][38]. Although the mechanisms of NLRP3 inflammasome activation are still debated, it is widely believed that changes in the mitochondrial membrane potential, permeabilization of the outer mitochondrial membrane, and increased formation of mitochondrial ROS are crucial factors in inducing the cytosolic translocation of mitochondrial molecules such as cardiolipin and mitochondrial DNA, which are capable of activating the inflammasome [43][36]. Indeed, the overproduction of ROS and dysregulation of the redox signaling system result in oxidative stress that can lead to mitochondrial damage, induce mitochondrial DNA mutations, damage the respiratory chain, alter membrane permeability, and affect the Ca2+ homeostasis and mitochondrial defense systems [46][39]. Accumulating evidence suggests that oxidative stress and mitochondrial injury can result in cellular DNA damage, degradation of proteins and lipids, and the pathogenesis of neurodegenerative diseases [47,48][40][41]. Nevertheless, cells have an accurate endogenous antioxidant defense system that can maintain cellular redox homeostasis between ROS production and elimination to ensure normal cellular signaling and redox regulation (Figure 1) [49][42].
The nuclear factor erythroid 2–related factor 2 (Nrf2) is an emerging therapeutic target, since it is involved in cellular resistance to oxidants. In detail, Nrf2 controls the basal expression of genes coding for enzymes and proteins involved in antioxidant and detoxifying action, repair and removal of damaged proteins and organelles, the inflammatory response, and mitochondrial bioenergetics [50,51][43][44]. Additionally, malfunctioning mitochondria can be selectively removed through a conserved cellular recycling process known as mitochondrial autophagy or mitophagy. In fact, the efficient elimination of damaged mitochondria prevents activation of cell death pathways, protects against ROS overproduction, and maintains efficient ATP production [52][45]. Damaged mitochondria are swallowed into autophagic vesicles that subsequently transport them to lysosomes for destruction. Mitophagy is a strictly regulated process, modulated by mitochondrial fission and fusion proteins, BCL-2 (B-cell lymphoma 2) family proteins [53][46], and the PINK1/Parkin pathway [54][47]. As all other defense mechanisms fail, the neuron can orchestrate its own-destruction by activating an intrinsic suicide program, otherwise known as apoptosis [55][48] (Figure 1). The underlying mechanisms of apoptosis are very sophisticated and engage an energy-dependent cascade of molecular events. In addition, it can follow different molecular pathways, one of which is the intrinsic pathway that involves the mitochondria [56][49]. Indeed, these organelles represent the site where anti-apoptotic and pro-apoptotic proteins interact and the origin of signals that initiate the activation of caspases, the cysteine proteases capable of cleaving many cellular substrates to disrupt cellular contents [57][50].
It is therefore understood that mitochondrial integrity and homeostasis are prerequisites for proper cellular functioning, especially for neurons, which are polarized, complex cells with high energy demands [58][51]. Not surprisingly, neurons have the highest content of mitochondria compared to other cell populations. Mitochondrial functionality is essential for ensuring membrane excitability and performing the complex neurotransmission and plasticity processes.
Firstly, the mitochondria provide the energy needed to undertake a wide range of neuronal functions, such as the maintenance of resting membrane potential, the restoration of ionic balance after depolarization, the cycling of synaptic vesicles, and the transport of proteins and organelles from the soma to distal sites. Interestingly, the mitochondria in axons and dendrites have different morphologies, some are small and sparsely distributed in the former, whereas others are elongated and densely distributed in the latter. In addition, axonal and dendritic mitochondria differ in movement, metabolism, and responses to neuronal activity [59][52].
Secondly, several lines of evidence support the role of mitochondria in the mobilization and recycling of synaptic vesicles [60][53]. Neurotransmission is underpinned by endocytosis and the local filling of synaptic vesicles in the presynaptic terminal [61][54]. Furthermore, the mitochondria support synaptic activity through cytosolic calcium reabsorption, a critical buffering mechanism for establishing and maintaining synaptic activity and preventing neuronal toxicity and excitotoxicity. Moreover, mitochondrial calcium buffering appears to be necessary at the synapse, even when the neuron is at rest: synaptic terminals lacking mitochondria show a higher frequency of spontaneous release of neurotransmitter-containing vesicles [62][55].
Overall, the importance of the mitochondria in neurons is unequivocal. Therefore, mitochondrial dysfunction leads to a plethora of severe conditions, from impaired neuronal development to various neurodegenerative diseases.
3. Role of Mitochondria in Neurodegenerative Disease
A great deal of evidence shows the effects of mitochondrial dysfunction on degenerative diseases [63][56]. Diseases caused by mitochondrial alterations often show a neurodegenerative component involving the nervous system. Similarly, mitochondrial defects are frequently observed in tissue samples taken from patients with a neurodegenerative disorder. This evidence reflected the high-energy requirements for all biological processes [64][57]. The continuous increase in energy demand, mainly due to excess food consumed daily, the constant increase in energy required for thermoregulation, or the increase in energy needed to counteract the environmental toxicity caused by humans, leads to an overwork of the mitochondria, resulting in impaired bioenergy efficiency [64,65][57][58]. It was demonstrated that cells exposed to an opulent nutrient environment are inclined to have their mitochondria in a fragmented state. Moreover, the mitochondria observed in cells in malnourished conditions remain for a longer period of time in the associated state [66,67][59][60]. This portrays the fact that the mitochondria can change their architecture and, therefore, their bioenergy capacity according to external events.
A degenerative disease is regarded as a type of non-physiological condition whose origins in a tissue or organ worsen over time. Energy failure has been linked to degenerative processes such as cancer, aging, neuroendocrine disease, neurodegenerative disease, and inflammatory diseases. Likewise, results from many studies support the involvement of the mitochondria in neurodegenerative diseases, particularly PD [68,69,70,71,72][61][62][63][64][65]. In addition, studies relating to the MPTP toxin first highlighted the role of mitochondrial complex I dysfunction and neurodegeneration in PD [73][66]. In association with other commonly used toxins, such as rotenone and paraquat, MPTP offered a new insight into defining the effective role of mitochondrial bioenergy in neurodegeneration [74][67]. Indeed, more than five thousand manuscripts dealing with the association between Parkinson’s disease and mitochondrial alteration can be found on PubMed (Figure 2). In addition, due to studies on PD, particularly genetic Parkinsonism, all mitochondrial dysfunctions leading to cell death have been well defined [75][68]. The PINK1 gene mutation, responsible for an early onset of Parkinsonism, serves as a good example [76][69]. This gene codes for the mitochondrial protein, phosphatase, and tensin homolog serine/threonine-protein kinase 1 (PTEN-induced kinase 1) [77][70]. PTEN-proteins can protect cells against oxidative stress, proton chain dysfunction, and bioenergy failure [76,78][69][71]. The PINK1 gene has also been identified as an oncogene with tumor suppressor properties [77,79,80][70][72][73]. Additionally, the PINK1 protective role has been observed in many disorders characterized by progressive inflammation and neurodegeneration, such as Alzheimer’s disease, multiple sclerosis, amyotrophic lateral sclerosis, and HD [77][70]. In physiological conditions, PINK1 is translocated inside the mitochondria in its mature isoform with the aim to withstand the activity of the mitochondrial chain and to produce (at the level of complex I) the molecules of ATP showing the maximal bioenergetic efficiency (Figure 1). Moreover, PINK1 is also located in the inner mitochondrial membrane and can interact with the chaperone TRAP1 (also recognized as an interactor of the type 1 tumor necrosis factor receptor) to maintain important bioenergetics and proteostatic functions [77,81][70][74]. Cleaved PINK1 can interact with other chaperon proteins, and when it is located in the cytosol, it can activate the m-Tork/Atk pathway. In addition, PINK1 can mediate the phosphorylation of another important gene for PD: Parkin. The coupled action of PINK1 and Parkin may induce mitochondrial fusion in order to cause the elimination of dysfunctional mitochondria. Furthermore, PINK1 is also involved in the formation of an autophagosome complex by the activation of beclin protein and may regulate the apoptotic process [77][70]. The role of the PINK1 gene in PD was well investigated by the use of animal models and cellular cultures of human fibroblasts [82,83,84,85,86,87][75][76][77][78][79][80] (Table 1). These studies have confirmed the role of bioenergetic efficiency in the maintenance of a healthy state or the progression of neurodegeneration. However, the absence of PINK1 results in low bioenergetic efficiency and programmed cell death after each minor stress [86,88][79][81]. It is very interesting to note that PINK1 not only has a key role in degeneration, oncological degenerative processes, and neurodegenerative PD but also in the neurodegeneration found in HD. Parkinson’s disease (PD) and HD are two different neurological diseases involving the central nervous system. The symptomatology of both is quite similar (cognitive impairment, limb inflexibility, and problems walking or talking), but while PD results from a combination of genetic and environmental factors, HD is only an inherited genetic disease. Both pathologies have a common factor, which is the loss of bioenergetic efficiency [77,89][70][82].
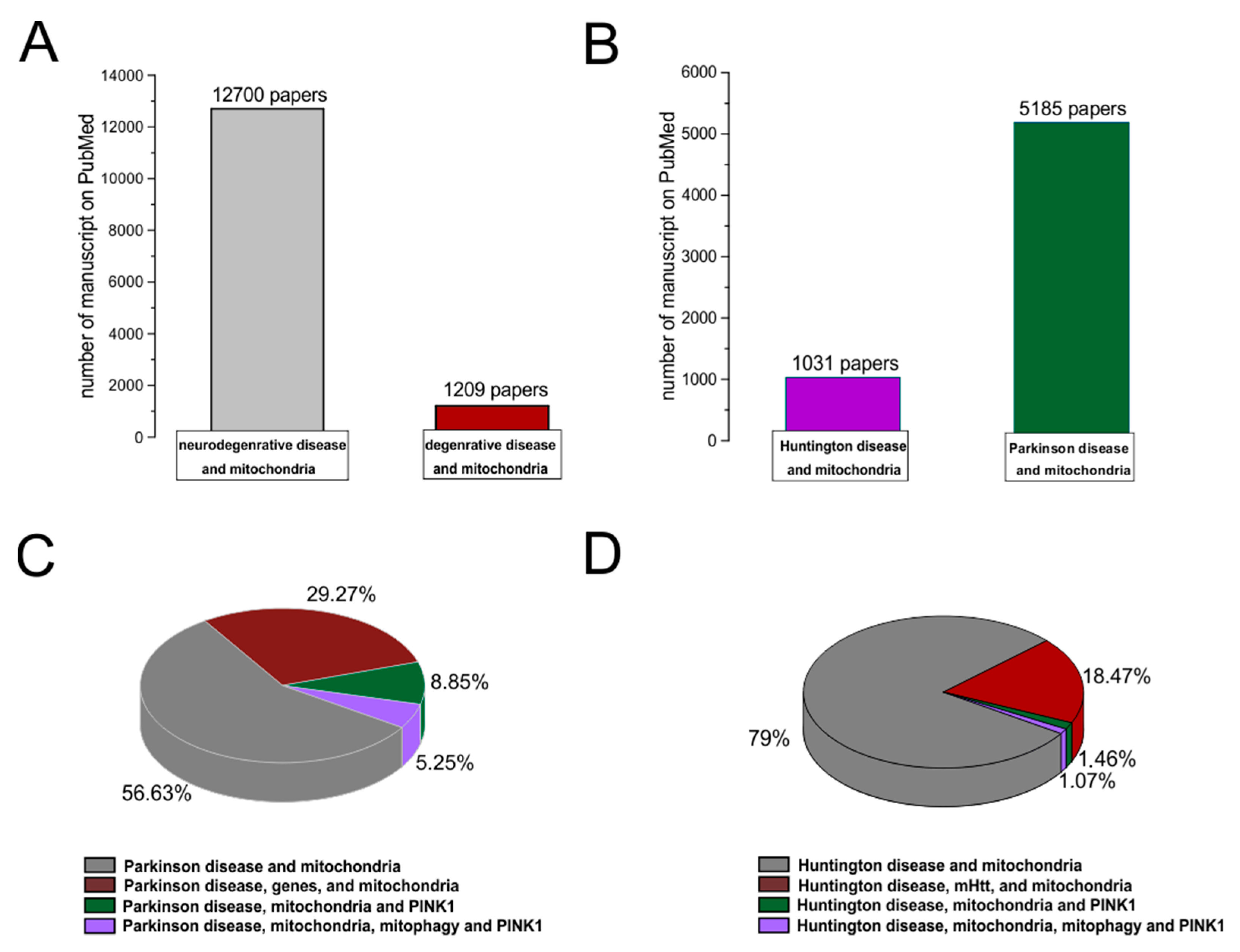
Figure 2. PubMed evidences of mitochondrial involvement in neuro-degenerative/degenerative diseases. Records were obtained from research articles published until January 2023. Article publications were obtained from the following PubMed searches: “Neurodegenerative disease and mitochondria; degenerative disease and mitochondria (A); HD and mitochondria; PD and mitochondria (B)”. (C,D) To realize the pies on sub-items, wthe researchers have selected “PD, genes, and mitochondria; PD, PINK1, and mitochondria; PD, PINK1, mitophagy, and mitochondria; HD, mHHT, and mitochondria; HD, mHHT, mitophagy, and mitochondria.
Table 1. Alteration of mitochondrial bioenergy function identified in genetic mouse models of PD and HD. The table summarizes the major mitochondrial changes found in murine models of PD and HD. The legend of the abbreviations is listed below.
| Mouse Model | Mitochondria Alteration | Molecules | References | |||||
|---|---|---|---|---|---|---|---|---|
| PD | α-synuclein A53T Mouse (Tg) | EN-mt | ↓ Drp1, ↓ Mfn1 | [90] | [83] | |||
| mtDNA damage | c-caspase-3 and p53 | [91] | [84] | |||||
| ↑ Mfn1, ↓ Mfn2 | [92] | [85] | ||||||
| A53T-hα-syn | mtUA | ↑ PRKAG2, ↑ TTR | [93] | [86] | ||||
| DE-autophagic/endocytic DA fibres | [93] | [86] | ||||||
| altered TCA cycle at striatal synapses | [93] | [86] | ||||||
| PINK1 KO mouse | ↑ number of larger mt | [94] | [87] | |||||
| ↓ respiratory complex I, II, III activity, age dependent; ↓ CAA and TCA cycle activity; ↑ protein oxidation | _ | [94] | [87] | |||||
| PINK1 KO rat | ↓ ATP production | ↑DRP1 | [95] | [88] | ||||
| defects complex I | ↑ O | 2 | consumption | [95] | [88] | |||
| increased complex II | [95] | [88] | ||||||
| bioinformatic analysis, PGC1A, PG1B, TFAM, GF1R, INSR, pathways were deactivated | [95] | [88] | ||||||
| DJ-1 mouse KO | ↓ aconitase | [96,97,98,99] | [89][90][91][92] | |||||
| activity; ↑ROS | ||||||||
| production | ||||||||
| ↑ Ca | [96,97,98,99] | [89][90][91][92] | ||||||
| ↑ GSH level and ↑ GSH/glutamate ↑ Glu | [96,97,98,99] | [89][90][91][92] | ||||||
| ↑ TCA cycle, H | 2 | O | 2 | consumption ↑ mitochondrial Trx activity, ↑ GSH and ↑ GSSG, ↑ GRX ↓ GR | [96,97,98,99] | [89][90][91][92] | ||
| Parkin mouse KO | DP, Cell Stress Chaperones and UPP components | [100] | [93] | |||||
| ↓ subunits of complexes I ↓subunits IV | ↓ peroxide reductases | [101] | [94] | |||||
| ↓ antioxidant capacity ↓ protein of lipid peroxidation | [101] | [94] | ||||||
| HD | R6/1 mouse | ↑ (ΔΨm) | ↑ Ca | 2+ | , ↑ NAD(P)H | [102] | [95] | |
| R6/2 mouse | ↑ OH(8)dG | [103] | [96] | |||||
| ↓ in NAA | [104] | [97] | ||||||
| ↑ glutamine ↑ glucose | [105] | [98] | ||||||
| ↑ creatine | [106] | [99] | ||||||
| ↑ GPC, ↑ glutamine and ↑ glutathione ↓ AA decreased at 8 weeks | [106] | [99] | ||||||
| reduction in mt complex IV activities (12 weeks) | ↑ iNOS and ↑nitrotyrosine | [107] | [100] | |||||
| ↓ aconitase cerebral cortex | ||||||||
| ↓ decrease in mitochondrial mass | synaptosomal ↑ ROS production and ↑ antioxidant in striatum | [108] | [101] | |||||
| YAC128 mouse | ↑ basal and maximal mitochondrial respiration | ↑ [64Cu]-ATSM | [109] | [102] | ||||
| ↑ ATP production, and ↑ complex II and III | [109] | [102] | ||||||
| ↑ oxygen consumption rate | [109] | [102] | ||||||
| ↓ Ca handling | [109] | [102] | ||||||
| YAC72 mouse | ↑ caspase-2 | [110] | [103] |
Parkinson disease (PD); Huntington disease (HD); decrease ↓; increase ↑; Mitochondrial membrane potential (ΔΨm); Mitochondria: mt; Transgenic animal model of human α-synuclein: α-synuclein A53T Mouse (Tg); AAV-mediated overexpression of α-synuclein: A53T-hα-syn; Enlarged neuronal mitochondria: EN-mt; Mitochondrial DNA damage: mtDNA damage; cleaved caspase-3: c-caspase-3; mitochondrial ultrastructural abnormalities: mtUA; disturbances exhibit autophagic: DE-autophagic; endocytic dopaminergic fibres: endocytic DA fibres: mitochondrial respiration or Krebs cycle: TCA cycle; cytosolic aconitase activity: CAA; proteins involved in detoxification: DP; stress-related chaperones: Cell Stress Chaperones; ubiquitin-proteasome pathway: UPP; 8-hydroxy-2-deoxyguanosine: OH(8)dG; N-acetylaspartate: NAA; Acetylaspartate: AA; Glycerophosphorylcholine: GPC; Inducible nitric oxide synthase: iNOS; aconitase cerebral cortex: Acon-CC; oxygen consumption rate: OCR.
Huntington disease is an incurable degenerative disorder caused by a mutation in the huntingtin gene, where the CAG sequence is excessively repeated. This mutation alters numerous cellular processes and leads to cell apoptosis. One important alteration is caused by the impairment of the mitochondrial metabolism. In a study conducted on the fly model, the formation of an abnormal ring-shaped mitochondria was observed; this particular shape was previously identified in mitophagy-blocked cells in which PINK1 overexpression was able to rescue the regular shape and function of the mitochondria. PINK1 over-expression was able to improve bioenergetic efficiency (increasing ATP levels) and rescue neuronal integrity in the adult drosophila model of HD [89][82]. HD is a degenerative pathology caused by the pathological expansion of CAG repeats in the Huntington gene, which codes for the Huntingtin protein. The gene is located on chromosome number 4 and is characterized by high levels of polymorphism. Unlike PD, HD has an age range of about 35–44 years and an estimated post-onset life span of 15/18 years. It is also characterized by an advanced motor disability, including chorea [111][104]. Enough evidence suggests that the mutation may cause an alteration in mitochondrial trafficking, an increase in oxidative stress, dyshomeostasis in intracellular calcium content, an alteration in bioenergetics, and an alteration in a respiratory mitochondrial chain [112,113,114,115][105][106][107][108]. Huntington’s disease, PD, and Alzheimer’s disease are three neurodegenerative diseases that have 37 common genes and about 40% of whose products act at the mitochondrial level [116][109]. These neurodegenerative diseases are coupled to a physiological degenerative process called aging or senescence that starts at the mitochondrial level and results in reduced bioenergetic efficiency [117][110]. Cellular senescence is characterized by heavy changes in cellular metabolism and an increase in pyruvate utilization that may produce differences in phosphorylation states, thereby increasing the activity of the mitochondrial pyruvate dehydrogenase complex and ROS production [118,119][111][112]. These features may lead to an impairment of cellular function that induces degeneration, as in the case of cancer or other neurodegenerative processes [120][113]. Moreover, recent data have demonstrated that key oncogenes and tumor suppressors modulate mitochondrial metabolism and dynamics. Indeed, different types of cancer result in more or less sensitivity to the modulation of mitochondrial function in the lesion caused by the tumor [121][114]. Therefore, the mitochondria play a cardinal role in many degenerative processes, which are mostly demonstrated in animal models.
References
- Osellame, L.D.; Blacker, T.S.; Duchen, M.R. Cellular and Molecular Mechanisms of Mitochondrial Function. Best Pract. Res. Clin. Endocrinol. Metab. 2012, 26, 711–723.
- Pizzorno, J. Mitochondria-Fundamental to Life and Health. Integr. Med. 2014, 13, 8–15.
- Bruce, A. Molecular Biology of the Cell; W. W. Norton & Company: New York, NY, USA, 2022.
- Wang, Y.; Xu, E.; Musich, P.R.; Lin, F. Mitochondrial Dysfunction in Neurodegenerative Diseases and the Potential Countermeasure. CNS Neurosci. Ther. 2019, 25, 816–824.
- Murali Mahadevan, H.; Hashemiaghdam, A.; Ashrafi, G.; Harbauer, A.B. Mitochondria in Neuronal Health: From Energy Metabolism to Parkinson’s Disease. Adv. Biol. 2021, 5, 2100663.
- Zhao, R.; Jiang, S.; Zhang, L.; Yu, Z. Mitochondrial Electron Transport Chain, ROS Generation and Uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15.
- Nolfi-Donegan, D.; Braganza, A.; Shiva, S. Mitochondrial Electron Transport Chain: Oxidative Phosphorylation, Oxidant Production, and Methods of Measurement. Redox Biol. 2020, 37, 101674.
- Trigo, D.; Avelar, C.; Fernandes, M.; Sá, J.; Cruz e Silva, O. Mitochondria, Energy, and Metabolism in Neuronal Health and Disease. FEBS Lett. 2022, 596, 1095–1110.
- McBride, H.M.; Neuspiel, M.; Wasiak, S. Mitochondria: More Than Just a Powerhouse. Curr. Biol. 2006, 16, R551–R560.
- Giorgi, C.; Agnoletto, C.; Bononi, A.; Bonora, M.; De Marchi, E.; Marchi, S.; Missiroli, S.; Patergnani, S.; Poletti, F.; Rimessi, A.; et al. Mitochondrial Calcium Homeostasis as Potential Target for Mitochondrial Medicine. Mitochondrion 2012, 12, 77–85.
- Pathak, T.; Trebak, M. Mitochondrial Ca2+ Signaling. Pharmacol. Ther. 2018, 192, 112–123.
- Shoshan-Barmatz, V.; Ben-Hail, D. VDAC, a Multi-Functional Mitochondrial Protein as a Pharmacological Target. Mitochondrion 2012, 12, 24–34.
- Rostovtseva, T.K.; Bezrukov, S.M.; Hoogerheide, D.P. Regulation of Mitochondrial Respiration by VDAC Is Enhanced by Membrane-Bound Inhibitors with Disordered Polyanionic C-Terminal Domains. Int. J. Mol. Sci. 2021, 22, 7358.
- Jung, H.; Kim, S.Y.; Canbakis Cecen, F.S.; Cho, Y.; Kwon, S.-K. Dysfunction of Mitochondrial Ca2+ Regulatory Machineries in Brain Aging and Neurodegenerative Diseases. Front. Cell Dev. Biol. 2020, 8, 599792.
- Sparagna, G.C.; Gunter, K.K.; Gunter, T.E. A System for Producing and Monitoring in Vitro Calcium Pulses Similar to Those Observed in Vivo. Anal. Biochem. 1994, 219, 96–103.
- Sparagna, G.C.; Gunter, K.K.; Sheu, S.-S.; Gunter, T.E. Mitochondrial Calcium Uptake from Physiological-Type Pulses of Calcium: A description of the rapid uptake mode. J. Biol. Chem. 1995, 270, 27510–27515.
- Grienberger, C.; Konnerth, A. Imaging Calcium in Neurons. Neuron 2012, 73, 862–885.
- Petersen, O.H. Calcium Signal Compartmentalization. Biol. Res. 2002, 35, 177–182.
- Laude, A.J.; Simpson, A.W.M. Compartmentalized Signalling: Ca2+ Compartments, Microdomains and the Many Facets of Ca2+ Signalling. FEBS J. 2009, 276, 1800–1816.
- Panda, S.; Behera, S.; Alam, M.F.; Syed, G.H. Endoplasmic Reticulum & Mitochondrial Calcium Homeostasis: The Interplay with Viruses. Mitochondrion 2021, 58, 227–242.
- Hirabayashi, Y.; Kwon, S.-K.; Paek, H.; Pernice, W.M.; Paul, M.A.; Lee, J.; Erfani, P.; Raczkowski, A.; Petrey, D.S.; Pon, L.A.; et al. ER-Mitochondria Tethering by PDZD8 Regulates Ca2+ Dynamics in Mammalian Neurons. Science 2017, 358, 623–630.
- Ashrafi, G.; de Juan-Sanz, J.; Farrell, R.J.; Ryan, T.A. Molecular Tuning of the Axonal Mitochondrial Ca2+ Uniporter Ensures Metabolic Flexibility of Neurotransmission. Neuron 2020, 105, 678–687.e5.
- Ryan, K.C.; Ashkavand, Z.; Norman, K.R. The Role of Mitochondrial Calcium Homeostasis in Alzheimer’s and Related Diseases. Int. J. Mol. Sci. 2020, 21, 9153.
- Görlach, A.; Bertram, K.; Hudecova, S.; Krizanova, O. Calcium and ROS: A Mutual Interplay. Redox Biol. 2015, 6, 260–271.
- Kuznetsov, A.V.; Margreiter, R.; Ausserlechner, M.J.; Hagenbuchner, J. The Complex Interplay between Mitochondria, ROS and Entire Cellular Metabolism. Antioxidants 2022, 11, 1995.
- Angelova, P.R.; Abramov, A.Y. Functional Role of Mitochondrial Reactive Oxygen Species in Physiology. Free Radic. Biol. Med. 2016, 100, 81–85.
- Angelova, P.R. Sources and Triggers of Oxidative Damage in Neurodegeneration. Free Radic. Biol. Med. 2021, 173, 52–63.
- Tait, S.W.G.; Green, D.R. Mitochondria and Cell Signalling. J. Cell Sci. 2012, 125 Pt 4, 807–815.
- Hordijk, P.L. Regulation of NADPH Oxidases: The Role of Rac Proteins. Circ. Res. 2006, 98, 453–462.
- Angajala, A.; Lim, S.; Phillips, J.B.; Kim, J.-H.; Yates, C.; You, Z.; Tan, M. Diverse Roles of Mitochondria in Immune Responses: Novel Insights Into Immuno-Metabolism. Front. Immunol. 2018, 9, 1605.
- Pearce, E.L.; Pearce, E.J. Metabolic Pathways in Immune Cell Activation and Quiescence. Immunity 2013, 38, 633–643.
- Breda, C.N.d.S.; Davanzo, G.G.; Basso, P.J.; Saraiva Câmara, N.O.; Moraes-Vieira, P.M.M. Mitochondria as Central Hub of the Immune System. Redox Biol. 2019, 26, 101255.
- Chen, Y.; Zhou, Z.; Min, W. Mitochondria, Oxidative Stress and Innate Immunity. Front. Physiol. 2018, 9, 1487.
- Chu, X.; Wu, S.; Raju, R. NLRX1 Regulation Following Acute Mitochondrial Injury. Front. Immunol. 2019, 10, 2431.
- Stokman, G.; Kors, L.; Bakker, P.J.; Rampanelli, E.; Claessen, N.; Teske, G.J.D.; Butter, L.; van Andel, H.; van den Bergh Weerman, M.A.; Larsen, P.W.B.; et al. NLRX1 Dampens Oxidative Stress and Apoptosis in Tissue Injury via Control of Mitochondrial Activity. J. Exp. Med. 2017, 214, 2405–2420.
- Liu, Q.; Zhang, D.; Hu, D.; Zhou, X.; Zhou, Y. The Role of Mitochondria in NLRP3 Inflammasome Activation. Mol. Immunol. 2018, 103, 115–124.
- Lamkanfi, M.; Kanneganti, T.-D. Nlrp3: An Immune Sensor of Cellular Stress and Infection. Int. J. Biochem. Cell Biol. 2010, 42, 792–795.
- He, Y.; Hara, H.; Núñez, G. Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem. Sci. 2016, 41, 1012–1021.
- Guo, C.; Sun, L.; Chen, X.; Zhang, D. Oxidative Stress, Mitochondrial Damage and Neurodegenerative Diseases. Neural Regen. Res. 2013, 8, 2003–2014.
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules 2019, 24, 1583.
- Behrouzi, A.; Kelley, M.R.; Fehrenbacher, J.C. Oxidative DNA Damage: A Role in Altering Neuronal Function. J. Cell. Signal. 2022, 3, 160–166.
- Poljsak, B.; Šuput, D.; Milisav, I. Achieving the Balance between ROS and Antioxidants: When to Use the Synthetic Antioxidants. Oxid. Med. Cell. Longev. 2013, 2013, 956792.
- Ma, Q. Role of Nrf2 in Oxidative Stress and Toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426.
- Dinkova-Kostova, A.T.; Kostov, R.V.; Kazantsev, A.G. The Role of Nrf2 Signaling in Counteracting Neurodegenerative Diseases. FEBS J. 2018, 285, 3576–3590.
- Thomas, R.L.; Gustafsson, A.B. Mitochondrial Autophagy—An Essential Quality Control Mechanism for Myocardial Homeostasis. Circ. J. Off. J. Jpn. Circ. Soc. 2013, 77, 2449–2454.
- Pattingre, S.; Tassa, A.; Qu, X.; Garuti, R.; Liang, X.H.; Mizushima, N.; Packer, M.; Schneider, M.D.; Levine, B. Bcl-2 Antiapoptotic Proteins Inhibit Beclin 1-Dependent Autophagy. Cell 2005, 122, 927–939.
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.-F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 Is Selectively Stabilized on Impaired Mitochondria to Activate Parkin. PLoS Biol. 2010, 8, e1000298.
- Elmore, S. Apoptosis: A Review of Programmed Cell Death. Toxicol. Pathol. 2007, 35, 495–516.
- Suen, D.-F.; Norris, K.L.; Youle, R.J. Mitochondrial Dynamics and Apoptosis. Genes Dev. 2008, 22, 1577–1590.
- Wang, C.; Youle, R.J. The Role of Mitochondria in Apoptosis. Annu. Rev. Genet. 2009, 43, 95–118.
- Sheng, Z.-H. The Interplay of Axonal Energy Homeostasis and Mitochondrial Trafficking and Anchoring. Trends Cell Biol. 2017, 27, 403–416.
- Seager, R.; Lee, L.; Henley, J.M.; Wilkinson, K.A. Mechanisms and Roles of Mitochondrial Localisation and Dynamics in Neuronal Function. Neuronal Signal. 2020, 4, NS20200008.
- Vos, M.; Lauwers, E.; Verstreken, P. Synaptic Mitochondria in Synaptic Transmission and Organization of Vesicle Pools in Health and Disease. Front. Synaptic Neurosci. 2010, 2, 139.
- Chanaday, N.L.; Cousin, M.A.; Milosevic, I.; Watanabe, S.; Morgan, J.R. The Synaptic Vesicle Cycle Revisited: New Insights into the Modes and Mechanisms. J. Neurosci. 2019, 39, 8209–8216.
- Datta, S.; Jaiswal, M. Mitochondrial Calcium at the Synapse. Mitochondrion 2021, 59, 135–153.
- Orth, M.; Schapira, A.H. Mitochondria and Degenerative Disorders. Am. J. Med. Genet. 2001, 106, 27–36.
- Liesa, M.; Shirihai, O.S. Mitochondrial Dynamics in the Regulation of Nutrient Utilization and Energy Expenditure. Cell Metab. 2013, 17, 491–506.
- Zhang, P.N.; Zhou, M.Q.; Guo, J.; Zheng, H.J.; Tang, J.; Zhang, C.; Liu, Y.N.; Liu, W.J.; Wang, Y.X. Mitochondrial Dysfunction and Diabetic Nephropathy: Nontraditional Therapeutic Opportunities. J. Diabetes Res. 2021, 2021, 1010268.
- Molina, A.J.A.; Wikstrom, J.D.; Stiles, L.; Las, G.; Mohamed, H.; Elorza, A.; Walzer, G.; Twig, G.; Katz, S.; Corkey, B.E.; et al. Mitochondrial Networking Protects Beta-Cells from Nutrient-Induced Apoptosis. Diabetes 2009, 58, 2303–2315.
- Gomes, L.C.; Di Benedetto, G.; Scorrano, L. During Autophagy Mitochondria Elongate, Are Spared from Degradation and Sustain Cell Viability. Nat. Cell Biol. 2011, 13, 589–598.
- Lewin, R. Trail of Ironies to Parkinson’s Disease. Science 1984, 224, 1083–1085.
- Hauser, D.N.; Hastings, T.G. Mitochondrial Dysfunction and Oxidative Stress in Parkinson’s Disease and Monogenic Parkinsonism. Neurobiol. Dis. 2013, 51, 35–42.
- Surmeier, D.J.; Obeso, J.A.; Halliday, G.M. Selective Neuronal Vulnerability in Parkinson Disease. Nat. Rev. Neurosci. 2017, 18, 101–113.
- Park, J.-S.; Davis, R.L.; Sue, C.M. Mitochondrial Dysfunction in Parkinson’s Disease: New Mechanistic Insights and Therapeutic Perspectives. Curr. Neurol. Neurosci. Rep. 2018, 18, 21.
- Magalhães, J.D.; Cardoso, S.M. Mitochondrial Signaling on Innate Immunity Activation in Parkinson Disease. Curr. Opin. Neurobiol. 2023, 78, 102664.
- Sagan, L. On the Origin of Mitosing Cells. J. Theor. Biol. 1967, 14, 225–274.
- Rossi, A.; Pizzo, P. Mitochondrial Bioenergetics and Neurodegeneration: A Paso Doble. Neural Regen. Res. 2021, 16, 686–687.
- Zhang, C.; Chen, S.; Li, X.; Xu, Q.; Lin, Y.; Lin, F.; Yuan, M.; Zi, Y.; Cai, J. Progress in Parkinson’s Disease Animal Models of Genetic Defects: Characteristics and Application. Biomed. Pharmacother. 2022, 155, 113768.
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.K.; Harvey, K.; Gispert, S.; Ali, Z.; Del Turco, D.; Bentivoglio, A.R.; Healy, D.G.; et al. Hereditary Early-Onset Parkinson’s Disease Caused by Mutations in PINK1. Science 2004, 304, 1158–1160.
- Arena, G.; Valente, E.M. PINK1 in the Limelight: Multiple Functions of an Eclectic Protein in Human Health and Disease. J. Pathol. 2017, 241, 251–263.
- Silvestri, L.; Caputo, V.; Bellacchio, E.; Atorino, L.; Dallapiccola, B.; Valente, E.M.; Casari, G. Mitochondrial Import and Enzymatic Activity of PINK1 Mutants Associated to Recessive Parkinsonism. Hum. Mol. Genet. 2005, 14, 3477–3492.
- Unoki, M.; Nakamura, Y. Growth-Suppressive Effects of BPOZ and EGR2, Two Genes Involved in the PTEN Signaling Pathway. Oncogene 2001, 20, 4457–4465.
- Garber, K. Parkinson’s Disease and Cancer: The Unexplored Connection. J. Natl. Cancer Inst. 2010, 102, 371–374.
- Masgras, I.; Laquatra, C.; Cannino, G.; Serapian, S.A.; Colombo, G.; Rasola, A. The Molecular Chaperone TRAP1 in Cancer: From the Basics of Biology to Pharmacological Targeting. Semin. Cancer Biol. 2021, 76, 45–53.
- Exner, N.; Treske, B.; Paquet, D.; Holmström, K.; Schiesling, C.; Gispert, S.; Carballo-Carbajal, I.; Berg, D.; Hoepken, H.-H.; Gasser, T.; et al. Loss-of-Function of Human PINK1 Results in Mitochondrial Pathology and Can Be Rescued by Parkin. J. Neurosci. 2007, 27, 12413–12418.
- Kitada, T.; Pisani, A.; Porter, D.R.; Yamaguchi, H.; Tscherter, A.; Martella, G.; Bonsi, P.; Zhang, C.; Pothos, E.N.; Shen, J. Impaired Dopamine Release and Synaptic Plasticity in the Striatum of PINK1-Deficient Mice. Proc. Natl. Acad. Sci. USA 2007, 104, 11441–11446.
- Martella, G.; Madeo, G.; Maltese, M.; Vanni, V.; Puglisi, F.; Ferraro, E.; Schirinzi, T.; Valente, E.M.; Bonanni, L.; Shen, J.; et al. Exposure to Low-Dose Rotenone Precipitates Synaptic Plasticity Alterations in PINK1 Heterozygous Knockout Mice. Neurobiol. Dis. 2016, 91, 21–36.
- Imbriani, P.; D’Angelo, V.; Platania, P.; Di Lazzaro, G.; Scalise, S.; Salimei, C.; El Atiallah, I.; Colona, V.L.; Mercuri, N.B.; Bonsi, P.; et al. Ischemic Injury Precipitates Neuronal Vulnerability in Parkinson’s Disease: Insights from PINK1 Mouse Model Study and Clinical Retrospective Data. Park. Relat. Disord. 2020, 74, 57–63.
- Imbriani, P.; Tassone, A.; Meringolo, M.; Ponterio, G.; Madeo, G.; Pisani, A.; Bonsi, P.; Martella, G. Loss of Non-Apoptotic Role of Caspase-3 in the PINK1 Mouse Model of Parkinson’s Disease. Int. J. Mol. Sci. 2019, 20, 3407.
- Brunelli, F.; Valente, E.M.; Arena, G. Mechanisms of Neurodegeneration in Parkinson’s Disease: Keep Neurons in the PINK1. Mech. Ageing Dev. 2020, 189, 111277.
- Zhi, L.; Qin, Q.; Muqeem, T.; Seifert, E.L.; Liu, W.; Zheng, S.; Li, C.; Zhang, H. Loss of PINK1 Causes Age-Dependent Decrease of Dopamine Release and Mitochondrial Dysfunction. Neurobiol. Aging 2019, 75, 1–10.
- Onyango, I.G.; Bennett, J.P.; Stokin, G.B. Regulation of Neuronal Bioenergetics as a Therapeutic Strategy in Neurodegenerative Diseases. Neural Regen. Res. 2021, 16, 1467–1482.
- Xie, W.; Chung, K.K.K. Alpha-Synuclein Impairs Normal Dynamics of Mitochondria in Cell and Animal Models of Parkinson’s Disease. J. Neurochem. 2012, 122, 404–414.
- Portz, P.; Lee, M.K. Changes in Drp1 Function and Mitochondrial Morphology Are Associated with the α-Synuclein Pathology in a Transgenic Mouse Model of Parkinson’s Disease. Cells 2021, 10, 885.
- Chinta, S.J.; Mallajosyula, J.K.; Rane, A.; Andersen, J.K. Mitochondrial α-Synuclein Accumulation Impairs Complex I Function in Dopaminergic Neurons and Results in Increased Mitophagy in Vivo. Neurosci. Lett. 2010, 486, 235–239.
- Metabolic Dysfunction in Parkinson’s Disease: Bioenergetics, Redox Homeostasis and Central Carbon Metabolism. Brain Res. Bull. 2017, 133, 12–30. Available online: https://pubmed.ncbi.nlm.nih.gov/28341600/ (accessed on 6 March 2023).
- Goldberg, J.A.; Guzman, J.N.; Estep, C.M.; Ilijic, E.; Kondapalli, J.; Sanchez-Padilla, J.; Surmeier, D.J. Calcium Entry Induces Mitochondrial Oxidant Stress in Vagal Neurons at Risk in Parkinson’s Disease. Nat. Neurosci. 2012, 15, 1414–1421.
- Li, Z.; Jo, J.; Jia, J.-M.; Lo, S.-C.; Whitcomb, D.J.; Jiao, S.; Cho, K.; Sheng, M. Caspase-3 Activation via Mitochondria Is Required for Long-Term Depression and AMPA Receptor Internalization. Cell 2010, 141, 859–871.
- Lopert, P.; Patel, M. Brain Mitochondria from DJ-1 Knockout Mice Show Increased Respiration-Dependent Hydrogen Peroxide Consumption. Redox Biol. 2014, 2, 667–672.
- Chen, W.; Liu, H.; Liu, S.; Kang, Y.; Nie, Z.; Lei, H. Altered Prefrontal Neurochemistry in the DJ-1 Knockout Mouse Model of Parkinson’s Disease: Complementary Semi-Quantitative Analyses with in Vivo Magnetic Resonance Spectroscopy and MALDI-MSI. Anal. Bioanal. Chem. 2022, 414, 7977–7987.
- Kirkinezos, I.G.; Moraes, C.T. Reactive Oxygen Species and Mitochondrial Diseases. Semin. Cell Dev. Biol. 2001, 12, 449–457.
- Dringen, R.; Gutterer, J.M.; Hirrlinger, J. Glutathione Metabolism in Brain Metabolic Interaction between Astrocytes and Neurons in the Defense against Reactive Oxygen Species. Eur. J. Biochem. 2000, 267, 4912–4916.
- Darios, F. Parkin Prevents Mitochondrial Swelling and Cytochrome c Release in Mitochondria-Dependent Cell Death. Hum. Mol. Genet. 2003, 12, 517–526.
- Goldberg, M.S.; Pisani, A.; Haburcak, M.; Vortherms, T.A.; Kitada, T.; Costa, C.; Tong, Y.; Martella, G.; Tscherter, A.; Martins, A.; et al. Nigrostriatal Dopaminergic Deficits and Hypokinesia Caused by Inactivation of the Familial Parkinsonism-Linked Gene DJ-1. Neuron 2005, 45, 489–496.
- Tkac, I.; Dubinsky, J.M.; Keene, C.D.; Gruetter, R.; Low, W.C. Neurochemical Changes in Huntington R6/2 Mouse Striatum Detected by in Vivo 1H NMR Spectroscopy. J. Neurochem. 2007, 100, 1397–1406.
- Ghiglieri, V.; Bagetta, V.; Calabresi, P.; Picconi, B. Functional Interactions within Striatal Microcircuit in Animal Models of Huntington’s Disease. Neuroscience 2012, 211, 165–184.
- Lopes, C.; Ferreira, I.L.; Maranga, C.; Beatriz, M.; Mota, S.I.; Sereno, J.; Castelhano, J.; Abrunhosa, A.; Oliveira, F.; De Rosa, M.; et al. Mitochondrial and Redox Modifications in Early Stages of Huntington’s Disease. Redox Biol. 2022, 56, 102424.
- Herbst, E.a.F.; Holloway, G.P. Exercise Training Normalizes Mitochondrial Respiratory Capacity within the Striatum of the R6/1 Model of Huntington’s Disease. Neuroscience 2015, 303, 515–523.
- Gardian, G.; Browne, S.E.; Choi, D.-K.; Klivenyi, P.; Gregorio, J.; Kubilus, J.K.; Ryu, H.; Langley, B.; Ratan, R.R.; Ferrante, R.J.; et al. Neuroprotective Effects of Phenylbutyrate in the N171-82Q Transgenic Mouse Model of Huntington’s Disease. J. Biol. Chem. 2005, 280, 556–563.
- Nithianantharajah, J.; Hannan, A.J. Dysregulation of Synaptic Proteins, Dendritic Spine Abnormalities and Pathological Plasticity of Synapses as Experience-Dependent Mediators of Cognitive and Psychiatric Symptoms in Huntington’s Disease. Neuroscience 2013, 251, 66–74.
- Ghiglieri, V.; Campanelli, F.; Marino, G.; Natale, G.; Picconi, B.; Calabresi, P. Corticostriatal Synaptic Plasticity Alterations in the R6/1 Transgenic Mouse Model of Huntington’s Disease. J. Neurosci. Res. 2019, 97, 1655–1664.
- Wellington, C.L.; Ellerby, L.M.; Hackam, A.S.; Margolis, R.L.; Trifiro, M.A.; Singaraja, R.; McCutcheon, K.; Salvesen, G.S.; Propp, S.S.; Bromm, M.; et al. Caspase Cleavage of Gene Products Associated with Triplet Expansion Disorders Generates Truncated Fragments Containing the Polyglutamine Tract. J. Biol. Chem. 1998, 273, 9158–9167.
- Schirinzi, T.; Salvatori, I.; Zenuni, H.; Grillo, P.; Valle, C.; Martella, G.; Mercuri, N.B.; Ferri, A. Pattern of Mitochondrial Respiration in Peripheral Blood Cells of Patients with Parkinson’s Disease. Int. J. Mol. Sci. 2022, 23, 10863.
- Anderson, K.E.; Marshall, F.J. Behavioral Symptoms Associated with Huntington’s Disease. Adv. Neurol. 2005, 96, 197–208.
- Caron, N.S.; Wright, G.E.; Hayden, M.R. Huntington Disease. In GeneReviews®; Adam, M.P., Everman, D.B., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993.
- Damiano, M.; Galvan, L.; Déglon, N.; Brouillet, E. Mitochondria in Huntington’s Disease. Biochim. Biophys. Acta 2010, 1802, 52–61.
- Rehman, M.U.; Sehar, N.; Dar, N.J.; Khan, A.; Arafah, A.; Rashid, S.; Rashid, S.M.; Ganaie, M.A. Mitochondrial Dysfunctions, Oxidative Stress and Neuroinflammation as Therapeutic Targets for Neurodegenerative Diseases: An Update on Current Advances and Impediments. Neurosci. Biobehav. Rev. 2023, 144, 104961.
- Jurcau, A.; Jurcau, C.M. Mitochondria in Huntington’s Disease: Implications in Pathogenesis and Mitochondrial-Targeted Therapeutic Strategies. Neural Regen. Res. 2023, 18, 1472–1477.
- Kawsar, M.; Taz, T.A.; Paul, B.K.; Ahmed, K.; Habib, M.A.; Bhuyian, T. Identification of Vital Regulatory Genes with Network Pathways among Huntington’s, Parkinson’s, and Alzheimer’s Diseases. Netw. Model. Anal. Health Inform. Bioinform. 2020, 9, 50.
- Li, Z.; Zhang, Z.; Ren, Y.; Wang, Y.; Fang, J.; Yue, H.; Ma, S.; Guan, F. Aging and Age-Related Diseases: From Mechanisms to Therapeutic Strategies. Biogerontology 2021, 22, 165–187.
- Quijano, C.; Cao, L.; Fergusson, M.M.; Romero, H.; Liu, J.; Gutkind, S.; Rovira, I.I.; Mohney, R.P.; Karoly, E.D.; Finkel, T. Oncogene-Induced Senescence Results in Marked Metabolic and Bioenergetic Alterations. Cell Cycle 2012, 11, 1383–1392.
- Sun, N.; Youle, R.J.; Finkel, T. The Mitochondrial Basis of Aging. Mol. Cell 2016, 61, 654–666.
- Moro, L. Mitochondrial Dysfunction in Aging and Cancer. J. Clin. Med. 2019, 8, 1983.
- Boland, M.L.; Chourasia, A.H.; Macleod, K.F. Mitochondrial Dysfunction in Cancer. Front. Oncol. 2013, 3, 292.
More