Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Catherine Yang and Version 1 by Angeliki Asimaki.
Arrhythmogenic cardiomyopathy (ACM) is a heart muscle disease associated with ventricular arrhythmias and sudden cardiac death (SCD). Pathologically, it is characterized by the gradual degeneration of cardiac myocytes (CMs) and their subsequent replacement by fat and fibrous tissue. The buccal mucosa can aid the diagnosis and understanding of this deadly disease.
- sudden cardiac death
- arrhythmogenic cardiomyopathy
- buccal mucosa
1. The Buccal Mucosa in Adult ACM
The buccal mucosa consists of flattened, squamous cells with small central nuclei and clearly identifiable edges. In normal subjects, these cells show strong membrane immunofluorescent signal for PG, DSP, Cx43 and plakophilin-1 (PKP1, Figure 1). PKP1 is one of the three plakophilin isoforms. It is primarily expressed in the upper epithelia. Conversely, PKP3 is primarily found in the lower epithelia, while PKP2 is the isoform expressed in the heart [59][1].
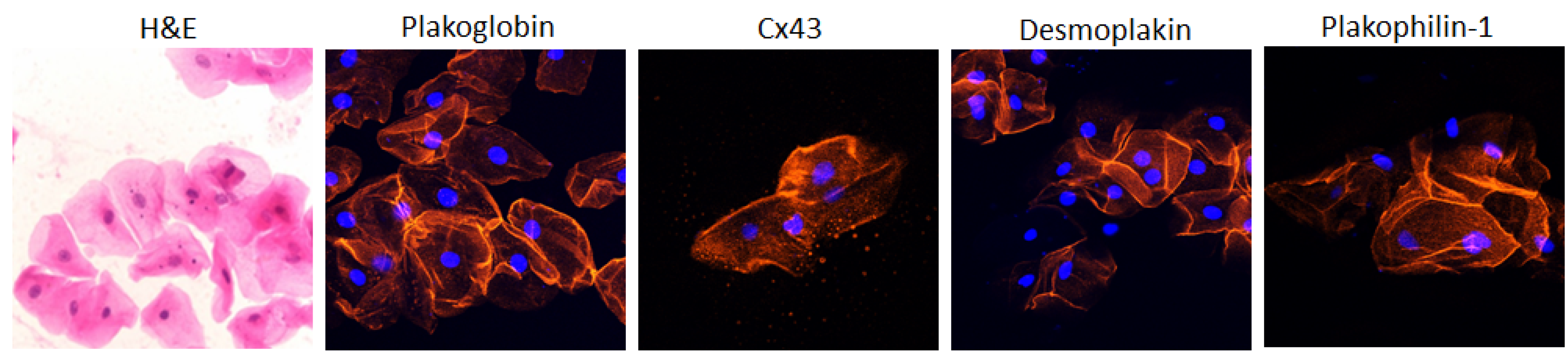
Figure 1. Representative confocal immunofluorescence images of buccal epithelial cells obtained from healthy controls. In the first image, cells are stained with hematoxylin and eosin (H&E). They show the typical squamous morphology; they have small central nuclei; and their edges are clearly identified. The remaining images show control buccal cells immunostained for plakoglobin, Cx43, desmoplakin and PKP1. The immunoreactive signal for all four proteins is strong and clearly localizes at the cell borders. Cell nuclei (blue) were stained with DAPI [59][1].
To determine whether the signal for these proteins is redistributed in the buccal mucosa in ACM, smears were obtained from a large number of patients bearing desmosomal gene mutations and subjected to immunocytochemistry. The membrane signal for PG was significantly depressed in 34/39 ACM subjects, while the membrane signal for Cx43 was virtually absent in 30/31 cases available. All proteins examined showed normal distribution in smears obtained from 40 control subjects. These were defined as individuals with no clinical signs or family history indicative of heart disease [59][1]. The signal for PKP1 was depressed in all patients bearing mutations in PKP2 but no patients with mutations in DSP, Dsc2, JUP or Dsg2. Conversely, the immunoreactive signal for DSP was reduced in all patients bearing mutations in Dsc2, Dsg2 or DSP but no patients with mutations in PKP2 or JUP.
As part of this study, 15 silent carriers were also sampled. These were individuals shown to carry a disease-causing mutation but without any clinical evidence of ACM. The signal for PG was depressed in 12/15 and the signal for Cx43 in 14/15 such carriers. Finally, the signal for PG was normal in smears from six patients diagnosed with different forms of heart disease (hypertrophic, dilated and ischemic cardiomyopathy), while the signal for Cx43 was depressed in all six sample sets [59][1]. Collectively, this study suggested a close relationship between the patterns of altered distribution of ID proteins in CMs and cell–cell junction proteins in buccal mucosa cells in patients with ACM. Interestingly, the study also showed that the localization of key proteins in buccal cells is correlated with the gene underlying the disease. Interestingly, PKP1-3 are isoforms of the same protein but are encoded by separate genes. In fact, those genes even reside on separate chromosomes. The fact that mutations in the PKP2-coding gene (which is only expressed in the heart) can alter the distribution of PKP1 (which is only found in the upper epithelia) suggests that the two genes are governed by yet unknown common regulatory pathways.
In a subsequent study, Driessen et al. obtained buccal smears from two patient groups in the Netherlands; the first group was diagnosed with ACM and found to carry desmosomal gene variants, while the second group was diagnosed with ACM associated with the R14del variant in the phospholamban gene (PLN). In the first group, immunoreactive signal for PG was significantly depressed in the buccal mucosa in both symptomatic ACM patients and asymptomatic mutation carriers compared to sex-matched controls (p = 0.002) [60][2]. When the amount of PG labeling was scored, a significant correlation was revealed between signal reduction in the buccal mucosa and the 2010 Task Force Criteria scores [6][3]; the most severely affected ACM patients showed the largest reduction in PG. Moreover, lower PG scores strongly correlated with the number of premature ventricular contractions on 24 h Holter monitoring (p = 0.02). However, these correlations were apparent only at a specific antibody dilution, highlighting the need for adherence to a specific research protocol for accurate data generation [60][2].
Plakoglobin distribution appeared normal and indistinguishable from controls in seven PLN-R14del carriers with a clinical diagnosis of DCM and in seven asymptomatic carriers of the mutation. Signal for PG was, however, significantly depressed in 3/3 R14del carriers with a clinical manifestation of ACM [60][2]. This is in agreement with the observations made in heart samples from R14del carriers, where a clinical diagnosis of ACM correlated with PG remodeling while DCM did not [30][4].
2. The Buccal Mucosa in Pediatric ACM
Up until recently, it had not been possible to correlate molecular changes, including redistribution of selected proteins, with the onset of disease in previously silent carriers. It had also not been possible to assess the effect of medication on similar molecular processes. As mentioned above, EMB procedures are difficult, risky and rarely performed on patients. Additionally, serial EMBs on a patient are even rarer. Obtaining a heart sample, however, from an individual who is a mutation carrier only, without showing the actual disease, is ethically unthinkable. This becomes even more of a science fiction scenario when the silent carriers are children. The use of buccal cells as a mirror of heart cells, however, has provided the opportunity to address these questions [61][5].
Key junctional proteins alter their distribution, shifting from the cell borders to the cell interior in buccal cells of children with a diagnosis of desmosomal ACM, as shown in adults [61][5]. Representative pictures are shown in Figure 2 [61][5].
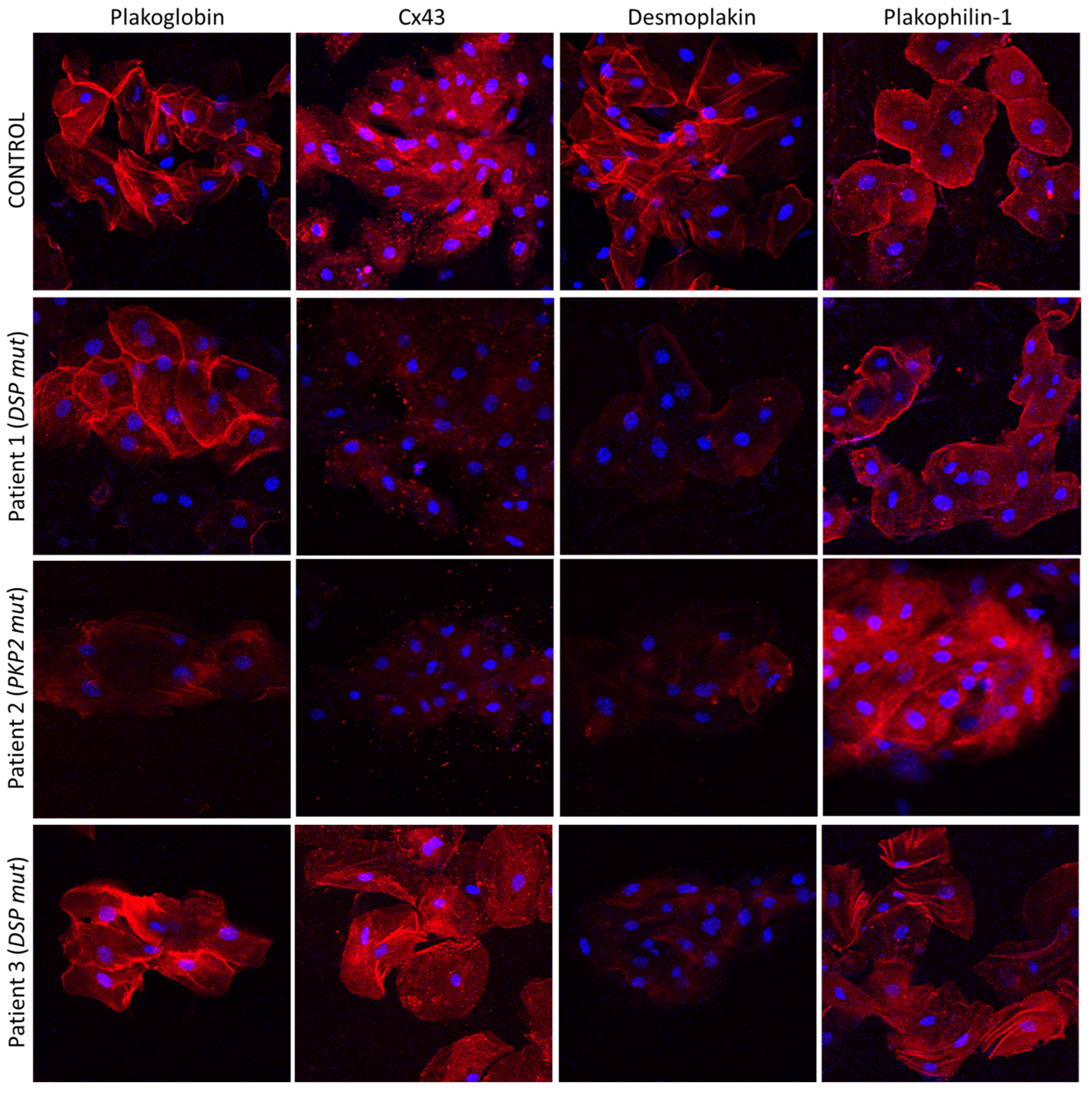
Figure 2. Confocal immunofluorescence images of buccal cells obtained from a healthy child (control) and three children with a clinical diagnosis of ACM. Patient 1 is bearing a pathogenic mutation in the DSP gene. He shows significantly reduced membrane signal for DSP and Cx43 compared to the control, but the distribution of PG and PKP1 appears to be normal. Patient 2 is carrying a pathogenic PKP2 mutation. She shows decreased localization of PG, DSP and Cx43 at the membranes, but the localization of PKP1 is indistinguishable from the control. Finally, Patient 3 carries a disease-causing mutation in DSP. DSP does not localize at the cell borders but PG, PKP1 and Cx43 do. The nuclei are stained blue with DAPI [61][5].
Interestingly, however, the localization of all examined proteins was normal and indistinguishable from controls in children carrying desmosomal gene variants without clinical manifestation of disease. The ease of buccal smear preparation allows for serial sampling, and hence, conduction of much needed molecular longitudinal studies. In the same study, Bueno-Beti et al. showed that, in addition to the initial protein set shifting at disease onset, further key protein distribution changes occur with clinical deterioration. Finally, the authors describe a case of an ACM patient initially showing reduced membrane localization of Cx43 [61][5]. An increased arrhythmic load prompted the initiation of a beta blocker. Holter monitoring several months later showed significant reduction in arrhythmias. A second set of buccal smears was obtained, and the localization of Cx43 now appeared membranous and indistinguishable from controls [61][5]. Representative images from this patient are shown in Figure 3. In summary, this study showed that analysis of buccal cells may be a straightforward, non-invasive and cost-effective way to confirm a diagnosis of ACM, to mark the clinical onset of disease, follow its progression over time and even assess whether therapeutic interventions show efficacy [61][5].
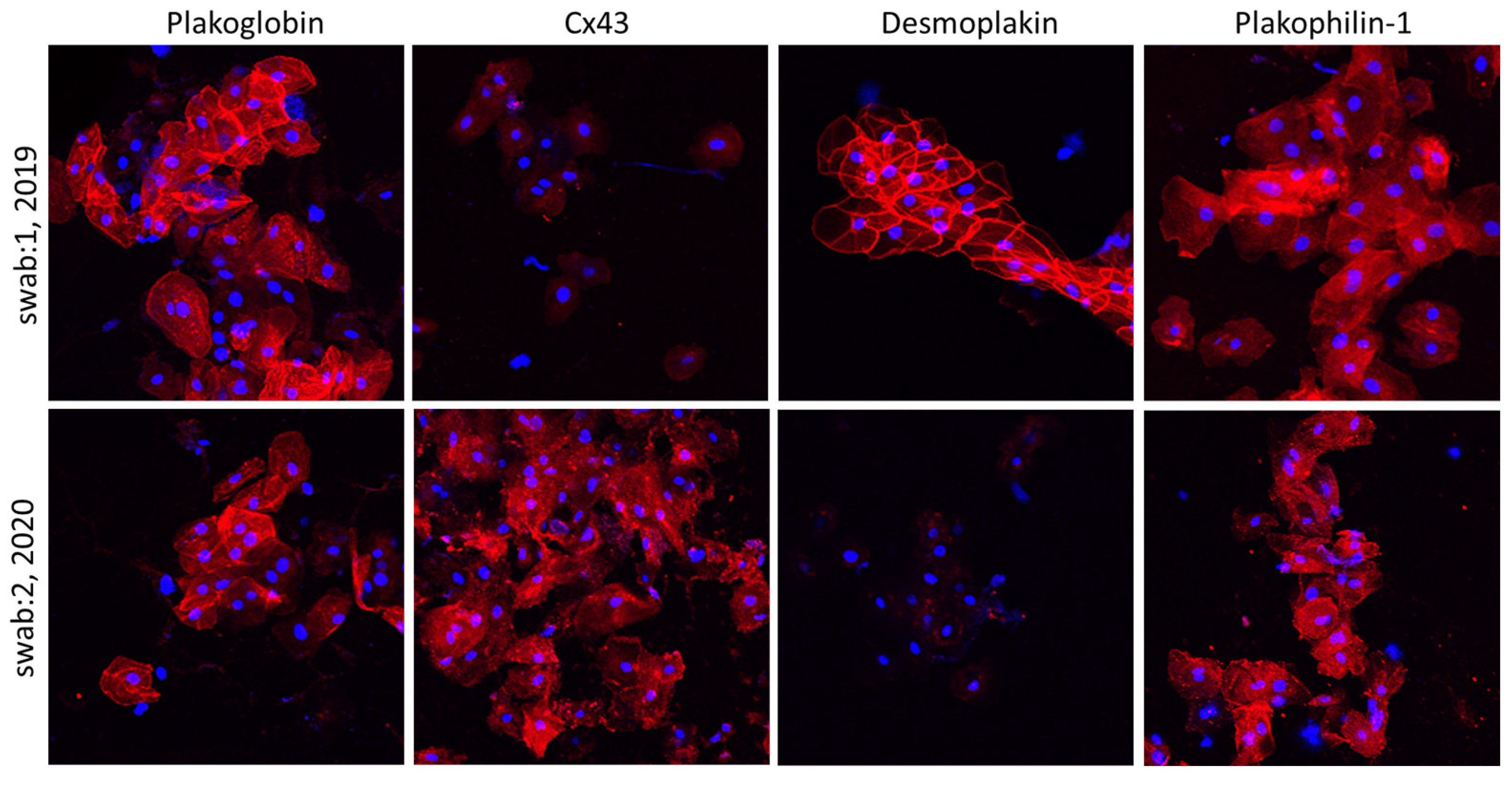
Figure 3. Confocal immunofluorescence images of two sets of buccal smears obtained from a child with a diagnosis of ACM bearing a DSP mutation. The first set of samples shows control localization for DSP, PG and PKP1, but membrane signal for Cx43 is virtually absent. In the second set of samples, obtained 18 months later, the immunoreactive signal for DSP appears to be missing from the membranes, while the distribution of Cx43 is now corrected to control-looking levels. Restoration of Cx43 signal correlated with a favorable response to beta blockers’ initiation [61][5].
References
- Asimaki, A.; Protonotarios, A.; James, C.A.; Chelko, S.P.; Tichnell, C.; Murray, B.; Tsatsopoulou, A.; Anastasakis, A.; Riele, A.T.; Kléber, A.G.; et al. Characterizing the Molecular Pathology of Arrhythmogenic Cardiomyopathy in Patient Buccal Mucosa Cells. Circ. Arrhythmia Electrophysiol. 2016, 9, e003688.
- Driessen, H.E.; van der Voorn, S.M.; Bourfiss, M.; van Lint, F.H.M.; Mirzad, F.; El Onsri, L.; Vos, M.A.; van Veen, T.A.B. Buccal mucosa cells as a potential diagnostic tool to study onset and progression of arrhythmogenic cardiomyopathy. Int. J. Mol. Sci. 2021, 23, 57.
- Marcus, F.I.; McKenna, W.J.; Sherrill, D.; Basso, C.; Bauce, B.; Bluemke, D.A.; Calkins, H.; Corrado, D.; Cox, M.G.; Daubert, J.P.; et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: Proposed Modification of the Task Force Criteria. Eur. Heart J. 2010, 121, 1533–1541.
- Van der Zwaag, P.A.; van Rijsingen, I.A.W.; Asimaki, A.; Jongbloed, J.D.H.; van Veldhuisen, D.J.; Wiesfeld, A.C.P.; Cox, M.; van Lochem, L.T.; de Boer, R.A.; Hofstra, R.M.W.; et al. Phospholamban R14del mutation in patients diagnosed with dilated cardiomyopathy or arrhythmogenic right ventricular cardiomyopathy: Evidence supporting the concept of arrhythmogenic cardiomyopathy. Eur. J. Heart Fail 2012, 14, 1199–1207.
- Bueno-Beti, C.; Field, E.; Tsatsopoulou, A.; Perry, G.; Sheppard, M.N.; Behr, E.R.; Saffitz, J.E.; Kaski, J.P.; Asimaki, A. Analysis of buccal mucosa as a prognostic tool in children with arrhythmogenic cardiomyopathy. Prog. Pediatr. Cardiol. 2022, 64, 101458.
More