More than 40 years ago, vesicle structures, similar to “cytoplasmic fragments” physiologically released, were identified in the cellular matrix. Their peculiarity was their ability to contain various materials, including ribosomes, which are involved in several pathological and physiological activities.These systems were defined as extracellular vesicles (EVs), and they include a wide variety of vesicles (from 30 nm to 5 μm) released from the plasma membrane (PM) of many different cell types into several bodily fluids. All EVs present a lipid bilayer membrane that surrounds a pool of genetic material, cytosolic proteins, or cellular debris. However, they significantly differ in terms of size, biogenesis, mechanisms, and function. Among these membrane vesicles, the most well-studied EVs are exosomes.
Exosomes have sizes ranging between 30 and 150 nm and represent a homogenous population of EVs released from cells when multivesicular bodies (MVBs) are fused with the membrane through inward budding in a highly regulated process. As natural carrier systems, exosomes present low immunogenicity, low toxicity, stability in the bloodstream, and efficient cell uptake due to their endogenous cellular tropism. Furthermore their ability to mediate intercellular communication allows their use as a promising therapeutic and diagnostic tool.
- extracellular vesicles
- miRNA
- biomarkers
- therapeutics
- tumor microenvironment
- glioma
- brain diseases
- intercellular communication
1. Introduction
The role of exosomes in cancer research has been rapidly growing over the last two decades. Cancer cells release a high number of exosomes containing many functional biomolecules in the extracellular space. EVs transfer proteins, receptors, and small RNAs that regulate both physiological and pathological processes. Moreover, the lipid bilayer membrane protects the exosome cargo from degradation in the bloodstream, allowing crossing different physiological barriers, such as the Blood–Brain Barrier (BBB) [1].
BBB is one of the most complex and selective barriers in the human organism. Its principal role is to preserve the homeostasis of the central nervous system and protect the brain parenchyma against the invasion of inflammatory mediators, which may interrupt its critical function. The BBB, together with pericytes, perivascular astrocytes, microglia, and neurons, forms a functional unit called the neurovascular unit. Interestingly, EVs regulate the communication between cells in short or long distances within the neurovascular unit. Furthermore, exosome cargo such as miRNAs, proteins, and other physiological compounds reflect different brain disease progression stages, allowing their use as a “window to the brain”[2][2] . Thus, engineered exosomes could represent a valid alternative to conventional drug delivery systems. However, more studies are required to identify the specific receptors involved in the transport as well as the mechanism of interaction with target cells.
2. Biogenesis of Exosomes: A Spontaneous Formation
The biogenesis and release of exosomes in the extracellular space initiate an endocytic pathway at the PM [3]. Despite the fact that this event is not entirely clarified, it begins with the formation and progressive accumulation of intraluminal vesicles (ILVs) in MVBs. These late vesicles elude the lysosomal digestive system and, after the fusion with the PM, are finally secreted into the extracellular space [4]. The physiological mechanism related to exosome formation and secretion is mediated via an Endosomal Sorting Complex Required for Transport (ESCRT)-dependent and/or ESCRT-independent pathway [5] (Figure 1).
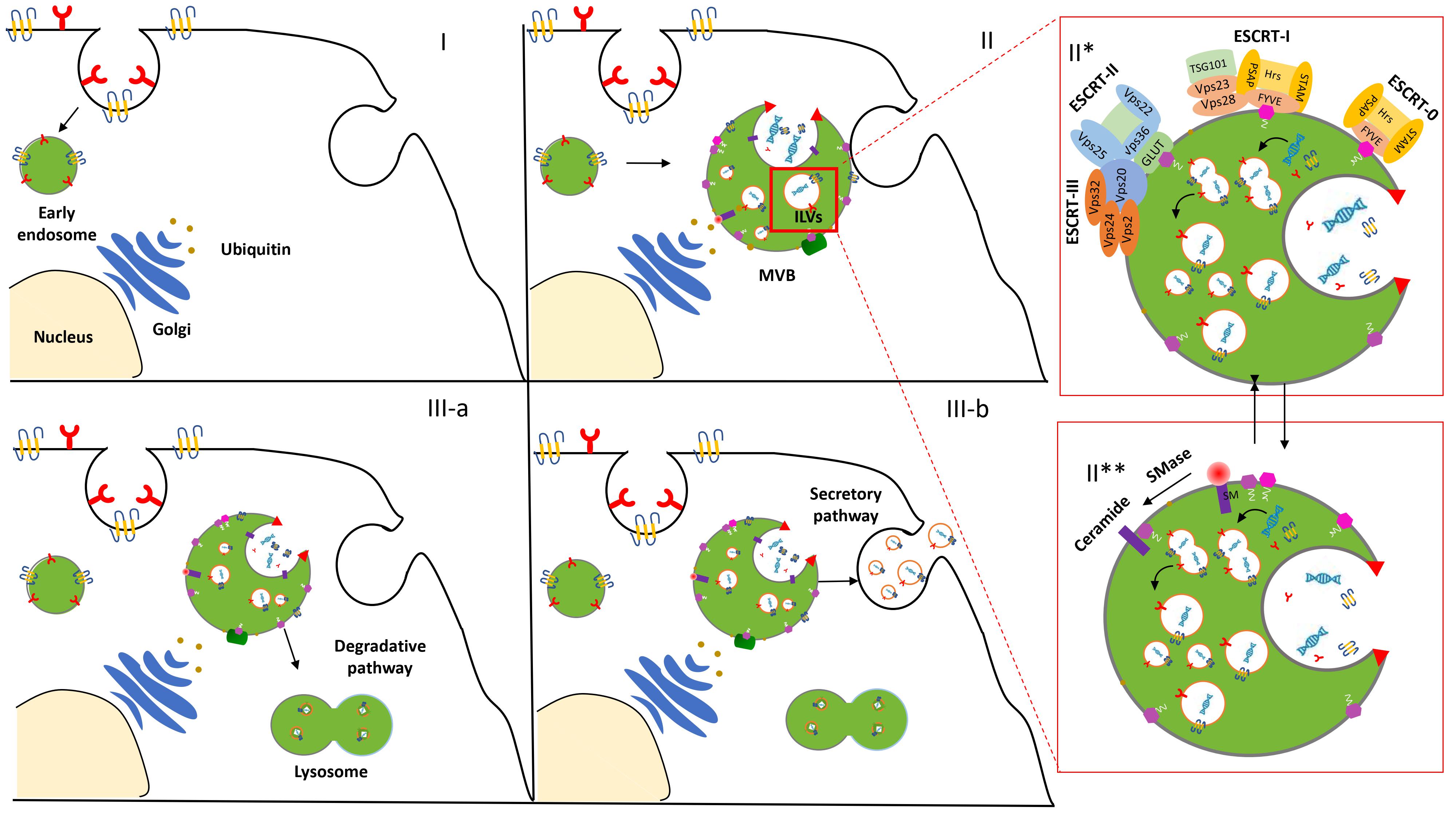
Figure 1. Biogenesis and secretion of exosomes. After the endocytosis of the plasma membrane, the transmembrane proteins are sorted into the vesicles that bud from the cellular membrane into “early endosomes” (I). The biogenesis of the exosome begins with the progressive formation and accumulation of ILVs inside MVBs (II). This process is mediated via an ESCRT-dependent (II*) and/or independent (II**) pathway. Then, the MVBs may follow a degradation pathway fusing with lysosomes or are destined to release the ILVs as exosomes to the extracellular space by exocytosis (III).
ESCRT machinery, conserved throughout eukaryotic and yeast cells, consists of four complexes—ESCRT-0, -I, -II, and -III—which act sequentially to bind and cluster ubiquitinylated proteins in the late endosome [6]. Structural and biochemical analyses of the upstream components and detailed studies of all the steps involved in the assembly and disassembly of the ESCRT complex contributed to its consideration as the main one implicated in EV biogenesis and clarified insights about EV formation and function.
However, several studies increased evidence of an additional ESCRT-independent pathways to form ILVs. Indeed, an unconventional pathway seems to be driven by the presence of certain lipids such as ceramides, as confirmed in the membrane trafficking of the proteolipid content in the oligodendroglial murine cell line [7]. These data provided evidence for an alternative pathway depending on raft-based microdomains that may contain high concentrations of sphingomyelina (SM).To date, it is legitimate to describe the presence of these two distinct processes as ESCRT-dependent or ESCRT-independent mechanisms. However, the activation of these alternative pathways is not fully elucidated and some aberrant ILV morphologies were observed while the early and late endosome remained differentiated Thus, we may hypothesize that the pathways are not entirely separated. They might work synergistically or influence each other. The cell type and/or cellular homeostasis could be an essential factor in controlling exosome secretion [8].
3. Mechanism of Interaction in the Biological Environment
The rising interest in EVs is due to their capacity to induce phenotypic changes in acceptor cells [9]. Exosomes play a key role in the systemic propagation of patho-physiological mechanisms, including development, homeostasis, and immune surveillance/pathogen response [10]. EVs’ internalization and regulatory properties depend on factors such as the maturation, physiological and environmental conditions of target cells, or even on the vesicular proteomic and lipidomic profile generally determined by the progenitor cell type [11]. The fact that EVs’ cargo reflects their tissue of origin is relevant, since cancer cells are known to produce greater numbers of EVs containing signaling molecules compared to healthy cells.
First of all, tumor-derived EVs at hypoxic conditions stimulate the neo-vascularization and propagation of the angiogenic phenotype to endothelial cells [12]. EVs’ angiogenic cargo includes a wide range of molecules such as tissue factors, cytokines, oncoproteins, sphingomyelin, and miRNAs. Pro-angiogenic factors include angiogenin, IL-6, IL-8, TIMP-1,and TIMP-2 that stimulate an angiogenic phenotype in normal brain endothelial cells and increase malignancy in a hypoxia-dependent manner [13]. Furthermore, tumor-derived EVs modulate the extracellular matrix through the proteolytic degradation of collagens, LNs, and fibronectin [14]. Matrix degradation has severe consequences on the tumor microenvironment, such as promoting host cell adhesion, motility, invasiveness, and apoptosis resistance. Hallal et al. [15] reported increased podosome formation and extracellular matrix degradation in astrocytes cultured with GBM exosomes. Interestingly, this phenomenon seems to strongly correlate with tumorigenesis through decreased p53 levels. As consequence, EVs modify neighboring astrocytes to induce tumor-supportive functions and, moreover, drive peritumoral astrocytes to become tumorigenic themselves. Conversely, in normal circumstances EVs are important in tissue homeostasis and organogenesis [16]. For example, platelet-derived EVs induce angiogenesis in vivo by facilitating the formation of endothelial capillaries [17]. Relevant studies show that exosomes produced by neurons, oligodendrocytes, astrocytes, and microglia have a key role in the protection and repair of brain tissue [18]. They could protect neurons by inhibiting neuronal apoptosis, modulate axon reconstruction and neurogenesis through vascular regeneration, and increase synaptic vesicle release [16].
Once released in the extracellular space, exosome internalization in a recipient cell occurs via two different mechanisms: direct interaction resulting in EV fusion with PM or endocytic uptake.
Endocytosis seems the principal pathway. It can involve multiple routes: the clathrin-dependent or independent pathway, the caveolin-mediated mechanism, micropinocytosis, phagocytosis, or lipid raft-mediated uptake [19]. Lipid composition is heavily involved in intracellular trafficking. The phosphatidylserine enrichment of oligodendrocyte-derived exosomes activated pinocytosis in a subset of microglia macrophages without antigen-presenting capability [20]. Additionally, the sphingolipids within the EV have an important role in binding and endocytosis, possibly through cholesterol-rich microdomains in dendritic cells [19]. Additionally, surface and cytoplasmic proteins anchored to the vesicle lipid bilayer membrane are involved in specific ligand-receptor type interactions. They include tetraspanins, TNF, TRAIL, FasL, integrins, or T cell immunoglobulin [21]. For example, tetraspanins are highly abundant on exosomes’ surfaces and notably have been shown to be involved in a number of processes, including vesicular and cellular fusion. The treatment of recipient cells with antibodies against the tetraspanins CD81 or CD9 can reduce the uptake of EVs by dendritic cells [22]. Cells over-expressing Tspan8 released EVs bearing a Tspan8-CD49d complex, the presence of which contributed to EV uptake by rat aortic endothelial cells [23]. Although there are several types of proteins capable of interacting specifically with a cellular target, none have been established as sufficient and necessary for EV internalization. Many EV subtypes share common surface proteins, and it is possible that one of them acts as a general ligand for a receptor, enabling vesicle internalization.
4. Exosomes in the Treatment of Glioma
In the last few decades, evidence about the role that exosomes secreted by healthy and tumor cells have in the growth and spread of such a complex environment suggested their use as a diagnostic and prognostic indicator of tumor progression, even for brain malignancies such as glioma (Table 1).
Table 1. Summary of the literature assessing exosomes as a drug delivery system and diagnostic biomarkers in in vitro and in vivo glioma models.
|
Cell Source |
Cargo |
Application |
Models |
|
Raw264.7 |
SPIONs/Curcumin/RGE peptide |
Imaging and anti-tumor therapy |
In vitro (U251) In vivo glioma mice model xenograft |
|
MSC |
miRNA-584 |
Anti-tumor miRNA therapy Inhibition glioma growth |
In vitro (U-87 MG) In vivo U-87 MG xenograft nude mouse mode |
|
MSC |
miR-199 |
Inhibition glioma growth Chemosensitivity |
In vitro (U251) Ex vivo immunohistochemistry tumor-bearing nude mice |
|
MSC |
miR-146b |
Anti-tumor miRNA therapy Inhibition glioma growth |
In vitro (9L glioma) Ex vivo rodent model (9L glioma) xenograft |
|
U-87 MG X12 cells |
miR-1 |
Anti-tumor miRNA therapy Inhibition glioma growth |
In vitro (U87 and X12 GBM) In vivo xenograft nude mouse model |
|
MSC |
anti-miR-9 |
Chemosensitivity |
In vitro (U-87 MG T98G) |
|
U-87 MG |
PTX/DXR |
Delivery anticancer drugs |
In vitro (U-87 MG) In vivo brain imaging of embryos zebrafish model |
|
Brain endothelial cell (bEND.3) |
|||
|
Mouse fibroblast cell line (L929) |
KLA peptide LDL/MTX |
Delivery of anticancer drug and therapeutic targeted peptides |
In vitro (U-87 MG) In vivo glioma mice xenograft |
|
MSC |
miR-124 |
Anti-tumor miRNA therapy Dysregulation of cellular metabolism |
In vitro (GSC26-28 GSC6-27) In vivo glioma mice xenograft |
|
Natural killer-92MI |
- |
Immunotherapy Inhibition Glioblastoma growth |
In vitro (U-87 MG) In vivo glioma mice xenograft |
|
CSF |
miR-21 |
Diagnostic biomarker |
- |
|
Serum |
miR-21/miR-222/miR-124-3p |
Diagnostic biomarker |
- |
|
CSF |
miR-21 miR-103, miR-24, and miR-125 |
Diagnostic biomarker |
- |
|
Serum |
miR-320/miR-574-3p/RNU6-1 |
Diagnostic biomarker Tumorigenesis factors |
- |
|
Serum |
miR-301a |
Diagnostic biomarker |
In vitro (H4) |
|
T98G cell line |
L1CAM |
Tumorigenesis factor |
Chick embryo brain tumor model |
|
Plasma |
CAV1 IL-8 |
Hypoxia-induced, proangiogenic proteins |
In vivo glioma mice xenograft |
|
Blood |
EGFR/EGFRvIII |
Diagnostic biomarker |
μNMR |
|
Blood |
PTRF |
Diagnostic biomarker |
In vitro (LN229 U-87 MG U251) In vivo mouse model xenograft |
Gliomas are the most frequent intrinsic tumors of the central nervous system and, by following the 2016 World Health Organization (WHO) update, encompass two principal subgroups: “nondiffuse gliomas”, showing a more circumscribed growth pattern (WHO grade I), and diffusely infiltrating gliomas (WHO grade II–IV), arising from glial cells or glial precursors [24]. Grade IV glioblastoma (GBM) is the most infiltrating, aggressive, and poorly treated brain tumor in adults. Despite the aggressive treatments used in GBM, such as surgical resection followed by radiotherapy and temozolomide (TMZ) therapy [25], limited drug penetration to the tumor sites and the rapid development of resistance to chemotherapy lead to poor prognosis. Thus, Exosomes’ ability as signaling in local and remote intercellular crosstalk enables them to deliver more efficiently macromolecular drugs, lipids, proteins, and genetic material such as miRNA siRNA to the brain. Further benefits can be achieved through exogenous or endogenous modification strategies (Figure 2).
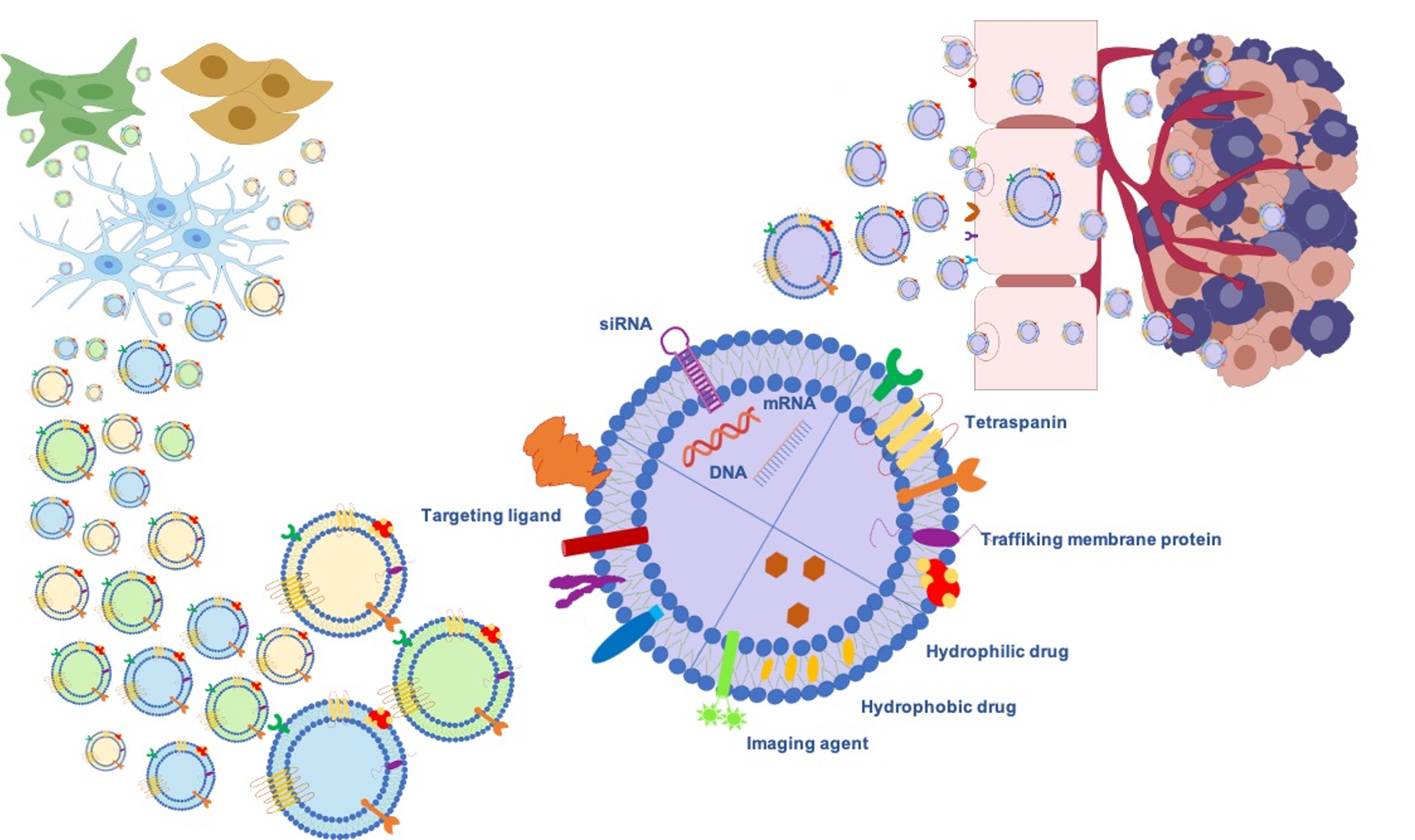
Figure 2. Exosomes as a promising biological nanoplatform for diagnosis and therapy in glioma treatment.
4.1 miRNAs Regulation as a Therapeutic Strategy
At date, exosomes derived from MSCs transfected with anti-tumor miRNAs have been found to be promising therapeutic tools for glioma therapy [26].
An example, Kim et al. [27] used exosome derived from MSCs transfected with miRNA-584. miRNA-584 acts as a tumor suppressor in some cancers and inhibits specifically glioma cells activity by binding to the 3′-UTR of CYP2J2. Interestingly, they demonstrated that MSCs exosomal miRNA-584 affects invasive ability of U-87 MG cells in vitro and decrease tumor mass weights in U87 MG xenograft nude mouse model. The glioma development could also be prevented by down-regulation of Arf GTPase- activating protein-2 (AGAP2), a target gene of microRNA-199a (miR-199a). In lines with this finding, Yu et al. [28] showed that miR-199a when delivered via MSCs-derived exosomes inhibiting in vitro U251 glioma cell proliferation, migration and invasion. Additionally, Katakowski et al. [29] tested MSC exosomes as a miRNA delivery vehicle in malignant glioma. Over-expressed miR-146b in MSC exosomes (M146-exo) were tested both in vitro and in vivo. Initially, they conducted an in vitro study on 9L cells and found out that, after 7 days, in vitro growth of M146-exotreated 9L cells was significantly less than normal MSC exosomes-treated control. Finally, to determine if M146-Exo had an anti-tumor effect in vivo, they administered M146-exo or M67-exo to Fischer rats bearing 9L gliosarcoma. One intra-tumor injection of M146-exo, 5 days after intracranial tumor xenograft implantation, led to a significant reduction in tumor volume at 10 days post-implant compared to control. Although MSC exosomes are the most commonly used therapeutic tool for miRNA transfection in glioma treatment, also tumoral exosomes could be applied. Indeed, it can not be excluded that the overexpression of specific tumoral biomolecules may further favor cellular exosomes communication with receiving cell and selectivity to the tumoral microenvironment. In this regard, Bronisz et al.[30] identified miR-1 deficiency as a contributor to glioma invasiveness and neovascularization and demonstrated that reintroduction of miR-1 into GBM exosomes through transfection of U-87 MG and X12 cells reverted paracrine-stimulated malignancy and microenvironmental remodeling by tumor. These findings support the hypothesis that miRNA replacement approaches have strong therapeutic potential and can be mediated by extracellular vesicles. In addition, they raise the possibility that modified tumor exosomes might be employed as biological Trojan horses to suppress tumor cells and their effect upon the brain microenvironment.
Reinforcing this point, it seems clear that miRNAs are often involved in the inhibition of tumor developmental processes. On the other hand, miRNA can behave not exclusively as tumor suppressors but also as oncogenes. Indeed, deregulation of microRNA expression has been observed in several cancers’ progression mechanisms, including GBM. Among these, Munoz et al. [31] focused on miR-9 molecules that have been shown to suppress the mesenchymal differentiation of GBM cells. They identified an increase of miR-9 concentration in TMZ-resistant GBM cells, involved in the expression of the drug efflux transporter P-glycoprotein. On this basis, they showed that reversed chemoresistance of GBM cells to TMZ occurred by targeting of anti-miR through MSCs. To block miR-9, they tested anti-miR-9-Exosome obtained from transfection with human bone marrow–derived MSC. Cell viability assay showed that the anti-miR-9-exosomes treatment enhances TMZ-induced cell death in U87 MG and T98G. Furthermore, transwell studies indicated that MSCs could communicate with cancer cells through gap junctional intercellular communication (GJIC) and also through secreted exosomes. Although further investigations have to be performed, this finding is relevant in the comprehension of exosomes and in vivo studies could confirm that the release of exosomes can affect GBM at a considerable distance from the MSCs.
4.2 Exosomes as Drug Delivery Systems
Finally, drug-loaded exosomes as drug delivery systems have been investigated by several authors. Although cellular packaging during EV biogenesis is a common and simple strategy, it involves the use of large amount of material and often has inefficient loading outcomes. The loading of EVs with therapeutic products after their isolation could represent a valid alternative. The simplest method is the passive incubation of isolated EVs with the therapeutic molecule, as reported by Yang et al. [32].
First of all, they highlighted the effect of the molecular characteristics of exosomes derived from the human brain neuronal glioblastoma-astrocytoma U-87 MG and the brain endothelial bEND.3 on their ability of interaction and the crossing of biological barriers. Their results demonstrated that bEND.3-derived exosomes allowed a higher internalization of the fluorescent marker in bEND.3 cells via an energy-dependent internalization process (cell uptake studies were performed both at 37 °C and 4 °C). Moreover, this active process was assumed to be receptor-mediated endocytosis by CD63 tetraspanins transmembrane proteins that are overexpressed in brain endothelial cells. Reinforcing this point, they reported the use of both U-87 MG and bEND.3 exosomes to deliver paclitaxel (PTX) or doxorubicin (DXR) across the BBB in a zebrafish model of brain tumor employing U-87 MG glioma. Freely administered DXR and PTX are not able to cross the BBB while the vesicles-packaged tool facilitated drug delivery across the BBB, reducing tumor progression. Promising experiments showed the possibility of simultaneous exosome engineering through surface modification and drug loading for imaging and therapy in vitro and in vivo. Jia et al. [33] firstly loaded superparamagnetic iron oxide nanoparticles (SPIONs) and curcumin (Cur) into exosomes and then conjugated the exosome membrane with neuropilin-1-targeted peptide (RGERPPR, RGE) by click chemistry to obtain glioma-targeting exosomes with imaging and therapeutic functions. Furthermore, the engineering of exosomes by both drug loading and surface functionalization was also recently performed by Ye et al. [34]. They reported a double functionalization of methotrexate (MTX)-loaded EVs with both the targeting pro-apoptotic peptide, KLA, and the targeted low-density lipoprotein, LDL, for selective binding to the LDL receptor (LDLR) overexpressed on the BBB and GBM cell lines. Indeed, the role of KLA was highlighted by observation under confocal microscopy. EVs decorated with KLA and LDL (EVs-KLA-LDL) were incubated with U-87 MG glioma spheroids for 12 h to assess their penetrating ability. EVs modified with the targeting peptide had an increased uptake by U-87 MG cells as well as an augmented permeation capacity into tumor cells. Furthermore, ex vivo fluorescence studies of the brain performed after intravenous injections of DiR-labeled EVs or EVs-KLA-LDL confirmed that EVs-KLA-LDL crosses the BBB and penetrates the brain more efficiently than blank-EVs, which might be attributed to the interaction between the LDL peptide and the LDLR over-expressed at the BBB. Thus, the engineering of the EV surface prompts the process of membrane receptor-mediated internalization both in vitro and in vivo and provides a unique opportunity to deliver KLA and MTX to the U-87 MG glioma. To improve the BBB permeation, studies have focused not only on chemical modifications and genetic engineering. The application of a focused ultrasound system (FUS) produce a reversible and local disruption of BBB; Bai et al. [35] designed a drug delivery system that combines doxorubicin (Dox)-loaded Exos derived from macrophages (R-Exos) and blood serum (B-Exos) for glioma diagnostics and therapy with two FUS treatments. Importantly, through this combination, they demonstrated a visible regression of tumor growth in orthotopic gliomas and an extended survival time, leading to a significant improvement over free Dox and Exos-Dox treatments.
References
- Sarko, D. K. and C. E. McKinney. Exosomes: Origins and therapeutic potential for neurodegenerative disease. Frontiers in Neuroscience 11 (2017): 7.Sarko, D. K. and C. E. McKinney. "Exosomes: Origins and therapeutic potential for neurodegenerative disease." Frontiers in Neuroscience 11 (2017): 7.
- Shi, M., L. Sheng, T. Stewart, C. P. Zabetian and J. Zhang. New windows into the brain: Central nervous system-derived extracellular vesicles in blood. Progress in neurobiology 175 (2019): 96-106.Shi, M., L. Sheng, T. Stewart, C. P. Zabetian and J. Zhang. "New windows into the brain: Central nervous system-derived extracellular vesicles in blood." Progress in neurobiology 175 (2019): 96-106.
- Keller, S., M. P. Sanderson, A. Stoeck and P. Altevogt. Exosomes: From biogenesis and secretion to biological function. Immunology Letters 107 (2006): 102-08.Keller, S., M. P. Sanderson, A. Stoeck and P. Altevogt. "Exosomes: From biogenesis and secretion to biological function." Immunology Letters 107 (2006): 102-08.
- Kalra, H., G. P. C. Drummen and S. Mathivanan. Focus on extracellular vesicles: Introducing the next small big thing. International Journal of Molecular Sciences 17 (2016)Kalra, H., G. P. C. Drummen and S. Mathivanan. "Focus on extracellular vesicles: Introducing the next small big thing." International Journal of Molecular Sciences 17 (2016)
- Munson, P. and A. Shukla. Exosomes: Potential in cancer diagnosis and therapy. Medicines 2 (2015): 310-27.Munson, P. and A. Shukla. "Exosomes: Potential in cancer diagnosis and therapy." Medicines 2 (2015): 310-27.
- Raiborg, C. and H. Stenmark. The escrt machinery in endosomal sorting of ubiquitylated membrane proteins. Nature 458 (2009): 445-52.Raiborg, C. and H. Stenmark. "The escrt machinery in endosomal sorting of ubiquitylated membrane proteins." Nature 458 (2009): 445-52.
- Trajkovic, K., C. Hsu, S. Chiantia, L. Rajendran, D. Wenzel, F. Wieland, P. Schwille, B. Brugger and M. Simons. Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science 319 (2008): 1244-47.Trajkovic, K., C. Hsu, S. Chiantia, L. Rajendran, D. Wenzel, F. Wieland, P. Schwille, B. Brugger and M. Simons. "Ceramide triggers budding of exosome vesicles into multivesicular endosomes." Science 319 (2008): 1244-47.
- Hessvik, N. P. and A. Llorente. Current knowledge on exosome biogenesis and release. Cellular and Molecular Life Sciences 75 (2018): 193-208.Hessvik, N. P. and A. Llorente. "Current knowledge on exosome biogenesis and release." Cellular and Molecular Life Sciences 75 (2018): 193-208.
- Mathieu, M., L. Martin-Jaular, G. Lavieu and C. Thery. Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell-to-cell communication. Nature cell biology 21 (2019): 9-17.Mathieu, M., L. Martin-Jaular, G. Lavieu and C. Thery. "Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell-to-cell communication." Nature cell biology 21 (2019): 9-17.
- De Toro, J., L. Herschlik, C. Waldner and C. Mongini. Emerging roles of exosomes in normal and pathological conditions: New insights for diagnosis and therapeutic applications. Frontiers in immunology 6 (2015): 203.De Toro, J., L. Herschlik, C. Waldner and C. Mongini. "Emerging roles of exosomes in normal and pathological conditions: New insights for diagnosis and therapeutic applications." Frontiers in immunology 6 (2015): 203.
- Oggero, S., S. Austin-Williams and L. V. Norling. The contrasting role of extracellular vesicles in vascular inflammation and tissue repair. Frontiers in Pharmacology 10 (2019)Oggero, S., S. Austin-Williams and L. V. Norling. "The contrasting role of extracellular vesicles in vascular inflammation and tissue repair." Frontiers in Pharmacology 10 (2019)
- Tadokoro, H., T. Umezu, K. Ohyashiki, T. Hirano and J. H. Ohyashiki. Exosomes derived from hypoxic leukemia cells enhance tube formation in endothelial cells. Journal of Biological Chemistry 288 (2013): 34343-51.
- Skog, J., T. Würdinger, S. Van Rijn, D. H. Meijer, L. Gainche, W. T. Curry, B. S. Carter, A. M. Krichevsky and X. O. Breakefield. Glioblastoma microvesicles transport rna and proteins that promote tumour growth and provide diagnostic biomarkers. Nature cell biology 10 (2008): 1470-76.
- Mu, W., S. Rana and M. Zöller. Host matrix modulation by tumor exosomes promotes motility and invasiveness. Neoplasia 15 (2013): 875-IN4.
- Hallal, S., D. Mallawaaratchy, H. Wei, S. Ebrahimkhani, B. Stringer, B. Day, A. Boyd, G. Guillemin, M. Buckland and K. L. Kaufman. Extracellular vesicles released by glioblastoma cells stimulate normal astrocytes to acquire a tumor-supportive phenotype via p53 and myc signaling pathways. Molecular neurobiology 56 (2019): 4566-81.
- Kang, X., Z. Zuo, W. Hong, H. Tang and W. Geng. Progress of research on exosomes in the protection against ischemic brain injury. Frontiers in Neuroscience 13 (2019): 1149.
- Lopatina, T., S. Bruno, C. Tetta, N. Kalinina, M. Porta and G. Camussi. Platelet-derived growth factor regulates the secretion of extracellular vesicles by adipose mesenchymal stem cells and enhances their angiogenic potential. Cell Communication and Signaling 12 (2014): 1-12.
- Budnik, V., C. Ruiz-Cañada and F. Wendler. Extracellular vesicles round off communication in the nervous system. Nature Reviews Neuroscience 17 (2016): 160-72.
- Mulcahy, L. A., R. C. Pink and D. R. F. Carter. Routes and mechanisms of extracellular vesicle uptake. Journal of extracellular vesicles 3 (2014): 24641.
- Toda, Y., K. Takata, Y. Nakagawa, H. Kawakami, S. Fujioka, K. Kobayashi, Y. Hattori, Y. Kitamura, K. Akaji and E. Ashihara. Effective internalization of u251-mg-secreted exosomes into cancer cells and characterization of their lipid components. Biochemical and biophysical research communications 456 (2015): 768-73.
- Robbins, P. D. and A. E. Morelli. Regulation of immune responses by extracellular vesicles. Nature Reviews Immunology 14 (2014): 195-208.
- Morelli, A. E., A. T. Larregina, W. J. Shufesky, M. L. Sullivan, D. B. Stolz, G. D. Papworth, A. F. Zahorchak, A. J. Logar, Z. Wang and S. C. Watkins. Endocytosis, intracellular sorting, and processing of exosomes by dendritic cells. Blood 104 (2004): 3257-66.
- Hromada, C., S. Mühleder, J. Grillari, H. Redl and W. Holnthoner. Endothelial extracellular vesicles—promises and challenges. Frontiers in Physiology 8 (2017): 275.
- Wesseling, P. and D. Capper. Who 2016 classification of gliomas. Neuropathology and applied neurobiology 44 (2018): 139-50.
- Mannas, J. P., D. D. Lightner, S. R. DeFrates, T. Pittman and J. L. Villano. Long-term treatment with temozolomide in malignant glioma. Journal of Clinical Neuroscience 21 (2014): 121-23.
- Schwarzenbach, H. and P. B. Gahan. Microrna shuttle from cell-to-cell by exosomes and its impact in cancer. Non-coding RNA 5 (2019): 28.
- Kim, R., S. Lee, J. Lee, M. Kim, W. J. Kim, H. W. Lee, M. Y. Lee, J. Kim and W. Chang. Exosomes derived from microrna-584 transfected mesenchymal stem cells: Novel alternative therapeutic vehicles for cancer therapy. BMB reports 51 (2018): 406.
- Yu, L., S. Gui, Y. Liu, X. Qiu, G. Zhang, X. a. Zhang, J. Pan, J. Fan, S. Qi and B. Qiu. Exosomes derived from microrna-199a-overexpressing mesenchymal stem cells inhibit glioma progression by down-regulating agap2. Aging (Albany NY) 11 (2019): 5300.
- Katakowski, M., B. Buller, X. G. Zheng, Y. Lu, T. Rogers, O. Osobamiro, W. Shu, F. Jiang and M. Chopp. Exosomes from marrow stromal cells expressing mir-146b inhibit glioma growth. Cancer Letters 335 (2013): 201-04. 10.1016/j.canlet.2013.02.019.
- Bronisz, A., Y. Wang, M. O. Nowicki, P. Peruzzi, K. I. Ansari, D. Ogawa, L. Balaj, G. De Rienzo, M. Mineo and I. Nakano. Extracellular vesicles modulate the glioblastoma microenvironment via a tumor suppression signaling network directed by mir-1. Cancer research 74 (2014): 738-50.
- Munoz, J. L., S. A. Bliss, S. J. Greco, S. H. Ramkissoon, K. L. Ligon and P. Rameshwar. Delivery of functional anti-mir-9 by mesenchymal stem cell-derived exosomes to glioblastoma multiforme cells conferred chemosensitivity. Molecular Therapy-Nucleic Acids 2 (2013): 10.1038/mtna.2013.60.
- Yang, T. Z., P. Martin, B. Fogarty, A. Brown, K. Schurman, R. Phipps, V. P. Yin, P. Lockman and S. H. Bai. Exosome delivered anticancer drugs across the blood-brain barrier for brain cancer therapy in danio rerio. Pharmaceutical Research 32 (2015): 2003-14.
- Jia, G., Y. Han, Y. L. An, Y. A. Ding, C. He, X. H. Wang and Q. S. Tang. Nrp-1 targeted and cargo-loaded exosomes facilitate simultaneous imaging and therapy of glioma in vitro and in vivo. Biomaterials 178 (2018): 302-16.
- Ye, Z. L., T. Zhang, W. S. He, H. L. Jin, C. W. Liu, Z. Yang and J. H. Ren. Methotrexate-loaded extracellular vesicles functionalized with therapeutic and targeted peptides for the treatment of glioblastoma multiforme. Acs Applied Materials & Interfaces 10 (2018): 12341-50.
- Bai, L., Y. Liu, K. Guo, K. Zhang, Q. Liu, P. Wang and X. Wang. Ultrasound facilitates naturally equipped exosomes derived from macrophages and blood serum for orthotopic glioma treatment. ACS applied materials & interfaces 11 (2019): 14576-87.