Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Camila Xu and Version 1 by Tao Yang.
Osteoarthritis (OA) is characterized by the degradation of joint cartilage tissue, chronic local inflammation, and bone remodeling, which result in joint pain, stiffness, swelling, and restricted motion. Aging phenotypes are considered a manifestation of accumulated cellular damage and defective repair. This is particularly apparent in the primary cell type of the articular joint, the articular chondrocytes. Articular chondrocytes are constantly facing the challenge of stressors, including mechanical overloading, oxidation, DNA damage, proteostatic stress, and metabolic imbalance.
- mechanical overloading
- oxidative stress
- DNA damage
- proteostatic stress
1. Introduction
Osteoarthritis (OA) is characterized by the degradation of joint cartilage tissue, chronic local inflammation, and bone remodeling, which result in joint pain, stiffness, swelling, and restricted motion [1[1][2],2], with an increased prevalence of over 110% in the last 30 years [3]. OA is highly associated with age, metabolic condition, genetic predisposition, and a history of joint injury or overuse [1]. OA is currently incurable, and standard treatments are pain control or joint replacement [4]. The difficulty in OA treatment reflects the unique properties of the joint tissues, particularly the articular cartilage, which is composed of articular chondrocytes (ACs) and lacks blood and lymphatic vessels [5]. ACs produce and remodel the extracellular matrix (ECM) to maintain a smooth and elastic gliding surface to facilitate movement of and resistance to shocks to the joints [5]. Except for those ACs with progenitor-like properties located in the superficial layer of the articular cartilage, the majority of ACs are long-lived postmitotic cells that are rarely replaced [6]. Over time, ACs accumulate stress-related damage, mostly resulting from mechanical overloading [7], oxidative stress [8], DNA damage, proteostatic stress, and metabolic imbalance. These stressors trigger AC homeostatic defects, including abnormal chondrocyte differentiation [9], senescence and apoptosis [8], a decline in ECM production [10] and an increase in ECM degradation and widespread inflammation [11], leading to OA. Taken together with the associated risk factors, stress acts as a major mediator between these factors and OA pathogenesis (Figure 1).
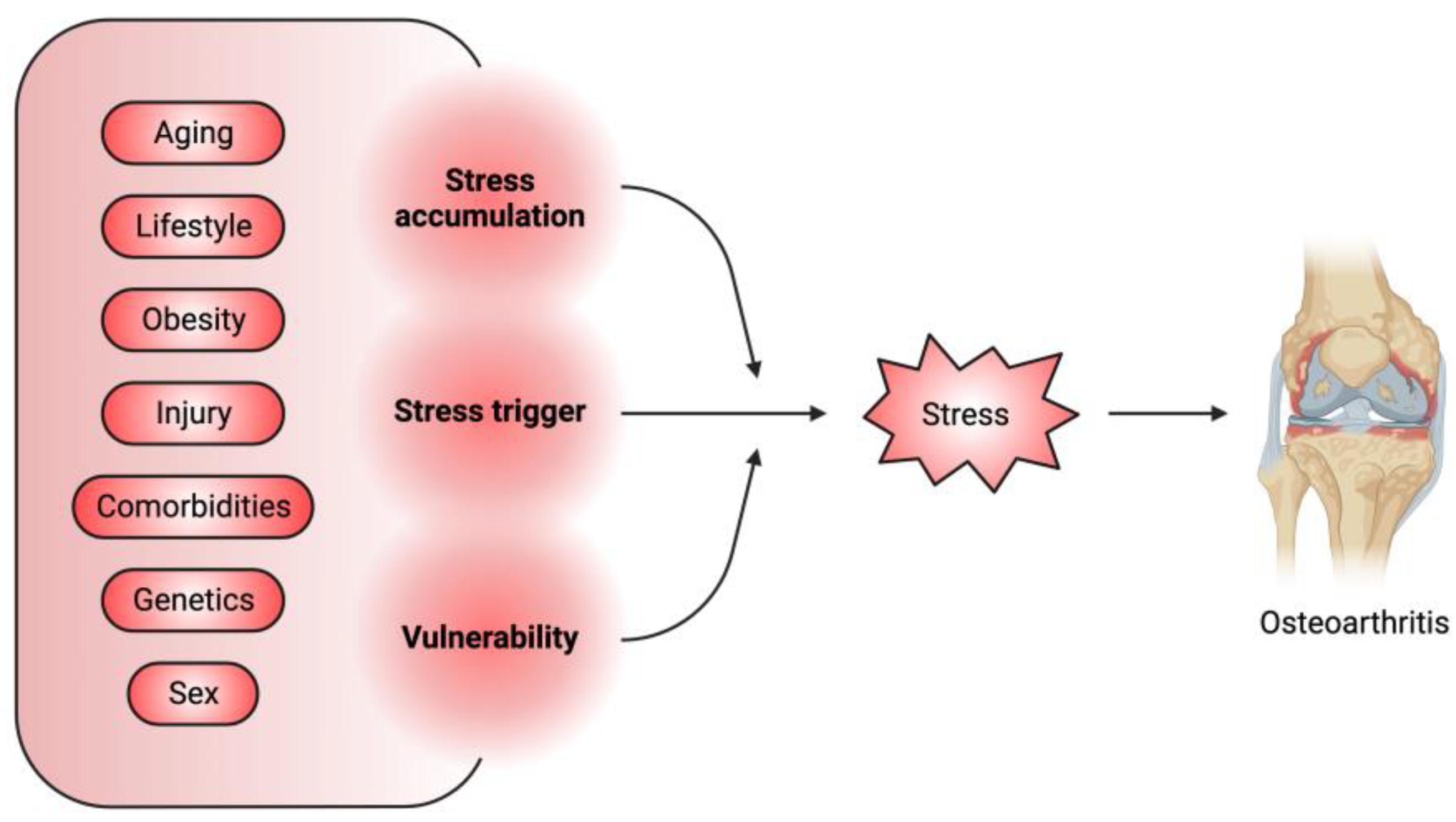
Figure 1. Stress is a major mediator between associated OA risk factors and pathogenesis. The OA-associated risk factors can serve as the initiator of stress or can promote the accumulation of stress-induced cellular damage or aggravate the vulnerability of joint tissues to these damages.
The long-lived ACs becoming senescent following the accumulation of cellular damage from stressors is supposed to promote AC survival by activating anti-apoptotic pathways, despite severely compromised functionality. Yet stressed ACs also produce the senescence-associated secretory phenotype (SASP), which consists of inflammatory and catabolic factors, such as tissue degrading enzymes [12], de facto functioning as the “contagious” root source of inflammation and tissue degradation in OA [13]. SASP produced from senescent ACs triggers a cascade of pathogenic events that lead to OA [14], supported by augmented senescence observed in cartilage in post-traumatic OA [15]. Overall, the presence of senescent ACs can act as a key component of a vicious loop of OA-related stressors: they can be both the source of new stressors and the products of others.
2. Primary Stressors in OA Pathogenesis
2.1. Mechanical Overloading
Normally, mechanical loading is necessary for joint health: by promoting cartilage thickness and ECM content, and by accelerating the exchange of fluid between the cartilage matrix and synovial cavity to supply the avascular articular cartilage with nutrients [19][16]. Cyclic loading also increases aggrecan biosynthesis in highly damaged and inflamed lesions, which can aid in repair [20][17]. In addition, proper level of mechanical loading is a source of anti-inflammatory processes [21,22][18][19]. However, excessive or cumulative mechanical stress, as a result of aging, joint injury, repetitive joint loading, joint misalignment, and obesity, damages joint tissues, in particular articular cartilage. In fact, introducing excessive mechanical stress to the joints is the main way to generate post-traumatic OA mouse models [23][20]. Chondrocytes sense mechanical loading through mechanosensing channels, cilia, integrins, and other elements of the focal adhesion complex, all of which convert loading information into the cell via intracellular signaling pathways. The TRPV4 Ca2+ preferred ion channel is a mechanosensitive channel present in ACs which increases the expression of ECM proteases under mechanical loading [24][21]. Chondrocyte-specific loss of TRPV4 alleviates the age-related OA phenotype in mice, suggesting that TRPV4-mediated mechanotransduction pathway can be a possible therapeutic target to treat aging-associated OA [25][22]. The mechanosensitive Ca2+ channels PIEZO1 and PIEZO2 are robustly expressed in ACs, and they potentiate the mechanically induced Ca2+ signals. A PIEZO-blocking peptide reduces chondrocyte apoptosis after mechanical injury in an explanted cartilage model, suggesting that PIEZO-mediated cartilage mechanotransduction may be a factor in OA pathogenesis and attenuating its activity may be a potential therapy for post-traumatic OA [26][23]. Increased intracellular Ca2+ concentration as a result of mechanical loading can trigger mitochondrial reactive oxygen species (ROS) production and cartilage degeneration [27][24]. Mechanical loading can also deform the cytoskeleton and cause damage to the mitochondria, leading to strain-mediated ROS release [27,28][24][25]. In a mouse model of OA, mechanical overloading reduces AC expression of superoxide dismutase 2 (SOD2), an enzyme that clears mitochondrial ROS; this exacerbates excessive mitochondrial superoxide formation and promotes AC apoptosis [29][26]. AC stressors do not act in isolation, and as is already apparent, mechanical overloading acts directly to increase oxidative stress. Oxidative stress in OA will be further elaborated below. Another arm of mechanical overloading that initiates OA development is the stimulation of defective joint repair, which requires the removal of damaged cartilage. This usually involves the interplay of integrins, kinase activations, augmented inflammatory factors, and cartilage catabolic enzymes. For example, mechanical overloading of chondrocytes activates integrin (αVβ3 and αVβ5)-mediated FAK and MAPK pathways to induce expression of tumor necrosis factor-α (TNFα), IL-1β, or NF-κB which results in MMP expression and ECM degradation [30][27]. In another example, mechanical loading leads to TNFα activation of NF-κB, MAPK, and c-Jun pathways and increases cartilage degrading MMP13 and ADAMT4/5 expression [31][28], ultimately inducing AC apoptosis [32][29]. Mechanical overloading-induced IL-1β further upregulates the expression of cartilage catabolic enzymes via MAPK pathways, amplifying inflammation by producing proinflammatory factors including COX-2, iNOS, PGE-2, and IL-6 [32][29]. During injury or overloading-induced cartilage remodeling, previously ECM-sequestered growth factors, such as TGFβ and FGF, are released [33][30]. TGFβ signaling acts as a double-edged sword in OA development. On the one hand, TGFβ blocks chondrocyte apoptosis and prevents cartilage degradation by promoting the production of cartilage matrix and lubricant while suppressing the expression of cartilage degrading enzymes [34,35,36,37][31][32][33][34]. TGFβ-SMAD2/3 signaling in ACs also has a protective role in AC degeneration and apoptosis following mechanical loading [38][35]. On the other hand, excessive mechanical load increases TGFβ activation in subchondral bone area, leading to marrow osteoid islet formation and enhanced angiogenesis, thus accelerating osteophyte formation and OA progression [39][36]. It was further determined that within subchondral bone, there is an uneven distribution of TGFβ. In areas of high mechanical loading, TGFβ expression is higher, but in areas of low mechanical stress, TGFβ expression is even lower than that in healthy tissue; both of these extremes are detrimental [40][37]. FGF2 expression appears to be beneficial in the joint, as FGF2 inhibits IL-1-induced aggrecanase production, thus alleviating OA progression [41][38]. Mechanical loading also alters the sensitivity of cartilage tissues to other signaling pathways. For example, tensile strain increases β-catenin levels and sensitizes WNT3A signaling-activated expression of cartilage catabolic proteases [42][39]. Moreover, chondrocytes under hydraulic pressure alter their expression of miR223, which promotes NF-κB signaling by suppressing the level of an NF-κB signaling inhibitor, IKKα [43][40]. As a downstream effector of many inflammatory cytokines, NF-κB signaling stimulates the expression of inflammatory factors as well as cartilage-degrading enzymes [44][41]. In addition, excessive mechanical loading was determined to activate the expression of Gremlin-1, which stimulates NF-κB signaling and promotes inflammation and oxidative stress in joints [45][42].2.2. Oxidative Stress
The oxidative pathway is activated by mechanical strain, but it also integrates the input from several other stressors. ROS are highly reactive free radicals containing oxygen molecules. ROS are a lethal weapon of phagocytic cells to attack and kill invaded pathogens and cancer cells. However, they also oxidize and damage proteins, DNA, and lipids [46][43]. Cells have an antioxidant system to scavenge ROS, primarily comprised of enzymes such as SOD and catalase, and small molecules, such as glutathione (GSH) and vitamin C [47,48][44][45]. Reduced expression of SIRT4, a deacetylase, was detected in OA cartilage; it leads to a decrease in SOD1, SOD2, and CAT expression, building up a more severe oxidative environment [49][46]. ROS production and oxidative stress are elevated in patients with OA, while OA cartilage has reduced expression of antioxidant enzymes. Together, these lead to an oxidative environment in OA tissue [50][47]. A major source of ROS is the mitochondrial electron transport chains, out of which 2–3% of O2 turns into O2−. O2− is also generated by NADPH oxidase, which is expressed in chondrocytes and also contributes to the OA-promoting oxidative environment that advances OA progression [51,52,53][48][49][50]. Finally, nitric oxide synthase (NOS) catalyzes the reaction that converts arginine into citrulline which produces the free radical, NO− [54][51]. Inducible NOS (iNOS) is upregulated in ACs by shear stress and proinflammatory factors, including IL-1β, NF-κB, and AP-1 [55][52]. iNOS loss in ACs prevents OA progression in mouse models, demonstrating that ROS production is a key factor in OA pathogenesis [56,57][53][54]. One major cellular consequence of oxidative stress is AC apoptosis. Given that ACs are the main cells responsible for cartilage matrix renewal and remodeling and that they are rarely replenished, their death fundamentally undermines the joint. NO is excessively produced from articular cartilage explants undergoing mechanical stress and is a potent inducer of chondrocyte apoptosis [58,59,60][55][56][57]. As a primary donor of ROS, mitochondria are also a victim of ROS and play a key role in oxidative stress-induced OA pathogenesis. ROS, if not cleared in time, can interrupt mitochondrial respiratory chain, reduce ATP production, and mutate mitochondrial DNA (mtDNA) [61,62,63][58][59][60]. H2O2 production and hyperoxidation of peroxiredoxin suppresses normal redox signaling, leading to an accumulation of H2O2 and disruption of physiological signaling. OA severity can be reduced in mice with increased expression of the antioxidant mitochondrial catalase [64][61]. While there is no significant change in expression of peroxiredoxin antioxidants in human ACs from older patients, the ACs are still more prone to hyperoxidation in comparison to ACs from younger patient tissue [65][62]. A recent study identified that oxidative stress-induced mitochondrial damage promotes mitochondrial double-strand RNA (mt-dsRNA) efflux. Chondrocytes exposed to H2O2, doxorubicin, or acute ionizing radiation (IR) induce cell senescence and elevate the cytosolic level of mt-dsRNAs. This further upregulates the expression of senescence-associated secretory phenotype (SASP), interferon β (IFN-β) and IFN-stimulated genes (ISGs), leading to inflammation through the activation of protein kinase R (PKR) and Toll-like receptor 3 (TLR3) pathways [66][63]. In addition, elevated levels of mt-dsRNA were detected in the synovial fluid of OA patients and cartilage of post-traumatic OA mice. In addition, removal of mt-dsRNA protects chondrocytes from those stresses, representing a promising strategy to treat OA [66][63]. Cartilage matrix synthesis is also severely affected in oxidative stress-induced OA progression. Endogenously produced ROS suppresses proteoglycan production in human articular cartilage [64][61]. In addition, introduction of oxidative stress in healthy donor ankle cartilage steers the normally pro-matrix-synthesis IGF1-PI3K/AKT pathway toward an anti-matrix-breakdown IGF1-MEK/ERK pathway [67][64]. Higher basal level of the IGF1-MEK/ERK pathway is also observed in human OA chondrocytes [67][64]. Moreover, inflammatory cytokines, such as IL-1, elevate the level of radicals [68][65], which further block the synthesis of proteoglycan, collagen type II, and aggrecan, as well as suppress chondrocyte progenitor migration to and proliferation at the injured foci [69][66].2.3. DNA Damage
DNA double-strand breaks and G/T or G/A transversions occur as a result of free radical oxidization of nucleoside bases [70][67], irradiation, genotoxins, and/or proinflammatory cytokines, leading to apoptosis and cellular senescence [71,72][68][69]. DNA damage increases linearly with age in human chondrocytes [73][70]. Furthermore, age-matched OA tissue has higher levels of DNA damage in comparison to healthy controls [73][70]. Elevated oxidative DNA damage in cartilage is positively correlated with OA symptom severity in a porcine OA model [74][71], and human cartilage explants irradiated to induce double-strand breaks and stimulated with exogenous mitogens, TGFβ and FGF [75][72], had accelerated chondrocyte senescence. Another study reported that the inflammatory factors, IL-1β and TNFα, increase free radicals in human chondrocytes, resulting in mitochondrial DNA (mtDNA) damage, impaired ATP production, and apoptosis [76][73]. In an additional study using porcine articular cartilage, mtDNA changes were more severe in the OA-associated chondrocytes than in healthy ACs, indicating that the OA-associated ACs are more sensitive to inflammatory signals in producing free radical production [77][74]. Disruption of metabolic pathways, such as the selenium pathway, by depleting selenophosphate synthetase 1 (SEPHS1) increases ROS levels and leads to DNA damage [78][75]. The Sephs1-deficient cells have reduced expression of those stress-related selenoproteins that act as oxidoreductases (including glutathione peroxidase 1 (GPX1) and methionine sulfoxide reductase B1 (MSRB1)) [79][76]. These deficient cells have stronger staining for γ-H2A.X (a marker of DNA damage) and higher expression of cartilage-degrading proteases ADAMTS5 and MMP13. In addition, SEPHS1 expression is decreased in human OA transcriptomes. These overall suggest that the selenium metabolic pathway is crucial for maintaining the health of ACs by decreasing ROS, and its loss/deficiency contributes to the progression of OA pathogenesis [79][76]. Expression of genes involved in DNA damage repair is changed in OA tissue. For example, the excision repair cross-complementation group 1 (ERCC1), an endonuclease required for DNA damage repair, is reduced in OA cartilage [80][77]. ERCC1 loss increases Mmp13 and suppresses Col2 expression, leading to degradation of ECM, while also promoting chondrocyte apoptosis and senescence [80][77]. In addition, the expression of cyclic GMP-AMP synthase (cGAS)-stimulator of interferon genes (STING), a component of the innate immune pathway that senses cytosolic DNA fragments derived from DNA damage, is markedly increased in OA tissues or IL-1β treated chondrocytes [81][78]. Gain of STING function suppresses ECM production, increases the expression of cartilage-degrading enzymes, promotes NF-κB signaling, and leads to chondrocyte apoptosis and senescence. Another recent work reported that irradiation-induced DNA damage stress synergizes with mitogenic stimuli by TGFβ and basic FGF to accelerate chondrocyte senescence in horse and human cartilage explants [75][72]. DNA damage impacts the expression of nuclear receptors in OA tissue. Estrogen receptor-α (ERα) decreases in human and mouse chondrocytes under Doxorubicin treatment-induced DNA damage [82][79]. In human OA tissues, the severely damaged regions have increased DNA damage in chondrocytes coupled with decreased ERα expression. Overexpression of ERα partially rescues the senescent phenotype of Doxorubicin-pretreated human ACs via suppressing NF-κB pathway [82][79]. This suggests that an estrogen-independent mechanism that regulates ERα is needed to maintain AC homeostasis [82,83][79][80].2.4. Proteostatic Stress
Proteostasis is the homeostatic state of a functional proteome. It is maintained by the proteostatic network that integrates protein synthesis, folding, trafficking and degradation in various cellular compartments [84][81]. Chondrocytes, as dedicated secretory cells, have robust protein synthesis activity and large endoplasmic reticula (ERs), so they are particularly vulnerable to proteostatic stress [16][82]. ER stress, a main type of proteostatic stress, occurs when the capacity for nascent protein folding in the ER becomes impaired. This is usually caused by protein overexpression, expression of misfolding-prone mutant proteins and dysregulated protein trafficking and degradation [16,85][82][83]. To handle ER stress, eukaryotic cells have evolved a conserved unfolded protein response (UPR), which relies on three unfolding protein sensing pathways: (1) IRE1/XBP1 pro-survival pathway; (2) ATF4 protective pathway; and (3) the PERK/ATF6/CHOP pro-apoptosis pathway. IRE1 is a transmembrane protein with both kinase and RNase activity. It can sense protein misfolding in the ER to become activated, then generates an XBP1 splicing variant, that encodes a stable form of the transcription factor XBP1 to promote the expression of UPR target genes. ER stress triggers ATF6 translocation from the ER to the Golgi apparatus, where ATF6 is cleaved and activated by S1P/S2P proteases. Cleaved ATF4 also enters the nucleus to activate UPR gene transcription. PERK, a serine/threonine ER protein kinase, phosphorylates and activates eIF2a to promote ATF4 mRNA translation. ATF4 induces the expression of a pro-apoptotic transcription factor, CHOP, leading to apoptosis of damaged cells [86][84]. Chondrocytes are sensitive to ER stress [87][85], and evidence of ER stress is present in human OA chondrocytes [87,88][85][86]. ER stress reduces transcription of cartilage matrix genes and promotes rat chondrocyte apoptosis [89][87]. Moreover, ER stress increases cartilage degradation via the expression of MMP13 in human chondrocytes [90][88], and reduces the XBP1-dependent protective UPR while enhancing the CHOP-dependent pro-apoptotic UPR, thus causing chondrocyte death [91][89]. The guiding of nascent protein folding or the degradation of misfolded proteins by chaperones is essential for minimizing protein aggregation and maintaining proteostasis. If the production or function of the molecular chaperones is impaired, accumulating misfolded proteins cause ER stress and induce UPR [84][81]. It has been observed that the expression of molecular chaperones, such as HSPA5 and BIP, is reduced in aging cynomolgus monkey articular cartilage, which sensitizes chondrocytes to ER stress, and ultimately leads to cell death [92][90]. In addition, siRNA knockdown of the chaperone, calnexin, in human chondrocytes increases the ER stress markers P-IRE1αm, XBP1, ATF4, and CHOP [92][90]. ER stress can also be induced by metabolic stress. For example, OA chondrocytes from diabetic OA patient cartilage and cultured in the presence of long-term high glucose diet express higher levels of the ER stress makers, GADD34, GRP78, and MANF, but suppress Col2 expression and cell proliferation [93][91]. In addition, mice fed with high-fat diet develop OA-like lesions exhibiting chondrocyte apoptosis. Treatment with 4-phenyl butyric acid, a chemical chaperone known to ease ER stress, alleviates the OA phenotypes in the high-fat-diet-fed mice [94][92]. These studies demonstrate that ER stress contributes to high-fat diet or obesity-induced OA, which can be targeted for OA therapeutics.2.5. Metabolic Stress
Metabolites are the source of energy and the building blocks of biomaterials for tissue homeostasis and renewal. However, chronic metabolite imbalance, or improper function in processing or using these metabolites, can bring forth stress to cells and ultimately lead to disease. High dietary fat consumption [95][93] and type 2 diabetes [95][93] can both accelerate the progression of knee OA. A meta-analysis integrating multiple studies showed that the risk for knee OA is increased by metabolic syndromes (MetS), defined as the presence of any three of the following components: abdominal obesity, hypertriglyceridemia, low high-density lipoprotein cholesterol; high blood pressure, and high fasting glucose [96][94]. These metabolic conditions accompany or induce systemic elevation of ROS and proinflammatory cytokines in OA [97][95]. A connection between high-fat diet, inflammation, and OA development has been established [98][96]. A number of nuclear receptors that regulate lipid metabolism contribute to OA progression by altering the production of cartilage ECM, cartilage degrading enzymes, antioxidants, and oxidative stress inducers [99][97]. In the case of high-fat diet, palmitate induces IL-1β-stimulated chondrocyte apoptosis [100][98]. Palmitate also upregulates COX2 and IL6 expression via TLR4 signaling and causes cartilage breakdown [100][98]. In addition, excessive free fatty acids released from lipid droplets can accumulate and induce apoptosis or metabolic disruption in ACs, defined as lipotoxicity [101][99]. Lipotoxicity and the pathological phenotypes of OA chondrocytes can be restrained by the protein kinase casein kinase 2 (PKCK2)-six-transmembrane protein of prostate 2 (STAMP2) and fat-specific protein 27 (FSP27) axis, which sequesters free fatty acids [101][99]. In contrast, a scarcity of lipids determines the chondrogenic fate of skeletal progenitors by activating the forkhead box O (FOXO) transcription factor, which activates SOX9 gene expression [102][100]. Besides its well-established function in initiating chondrogenesis, SOX9, in turn, suppresses oxidation of fatty acids, allowing chondrocytes to sustain an avascular microenvironment with low lipid supply [102][100]. Aberrant carbohydrate metabolism, which can be a result of alterations in dietary input, transport, and receptor usage, is also associated with pathological changes in chondrocytes. High-sucrose diet can recapitulate characteristics of early-stage OA, including chondrocyte hypertrophy and higher synovial cellularity independent of weight [103][101]. The high-sucrose diet also decreases antioxidant proteins, glycolysis-related enzymes, and the expression of genes related to mitochondria function [103][101]. Glucose is also a major energy source for chondrocytes, and its cellular entry requires the glucose transporter GLUT1. Glut1 deletion during development disrupts chondrocyte proliferation and hypertrophy resulting in a skeletal phenotype [104][102]. Notably, Glut1 expression is mediated by the BMP-mTORC1-HIF1α axis [104][102]. Extrapolating from the developmental context may provide new insights into AC homeostasis because related signaling pathways are found in OA pathogenesis [105][103]. Hyperglycemia and aging promote the accumulation of advanced glycation end products (AGE) as a result of a spontaneous reaction that adds a sugar group onto lipids, proteins, and ECM. AGEs trigger signaling via receptor for AGE (RAGE), which increases ROS and inflammation in articular joint tissues and promotes OA development [106,107][104][105]. Furthermore, the accumulation of intracellular AGEs induces ER stress and leads to CHOP-mediated chondrocyte apoptosis [108][106].References
- Palazzo, C.; Nguyen, C.; Lefevre-Colau, M.M.; Rannou, F.; Poiraudeau, S. Risk factors and burden of osteoarthritis. Ann. Phys. Rehabil. Med. 2016, 59, 134–138.
- Hunter, D.J.; Bierma-Zeinstra, S. Osteoarthritis. Lancet 2019, 393, 1745–1759.
- Long, H.; Liu, Q.; Yin, H.; Wang, K.; Diao, N.; Zhang, Y.; Lin, J.; Guo, A. Prevalence Trends of Site-Specific Osteoarthritis From 1990 to 2019: Findings from the Global Burden of Disease Study 2019. Arthritis Rheumatol. 2022, 74, 1172–1183.
- Grässel, S.; Muschter, D. Recent advances in the treatment of osteoarthritis. F1000Res 2020, 9, F1000 Faculty Rev-325.
- Sophia Fox, A.J.; Bedi, A.; Rodeo, S.A. The basic science of articular cartilage: Structure, composition, and function. Sport Health 2009, 1, 461–468.
- Jiang, Y.; Tuan, R.S. Origin and function of cartilage stem/progenitor cells in osteoarthritis. Nat. Rev. Rheumatol. 2015, 11, 206–212.
- Liu, Q.; Hu, X.; Zhang, X.; Duan, X.; Yang, P.; Zhao, F.; Ao, Y. Effects of mechanical stress on chondrocyte phenotype and chondrocyte extracellular matrix expression. Sci. Rep. 2016, 6, 37268.
- Brandl, A.; Hartmann, A.; Bechmann, V.; Graf, B.; Nerlich, M.; Angele, P. Oxidative stress induces senescence in chondrocytes. J. Orthop. Res. Off. Publ. Orthop. Res. Soc. 2011, 29, 1114–1120.
- Barbero, A.; Grogan, S.; Schafer, D.; Heberer, M.; Mainil-Varlet, P.; Martin, I. Age related changes in human articular chondrocyte yield, proliferation and post-expansion chondrogenic capacity. Osteoarthr. Cartil. 2004, 12, 476–484.
- Verbruggen, G.; Cornelissen, M.; Almqvist, K.F.; Wang, L.; Elewaut, D.; Broddelez, C.; de Ridder, L.; Veys, E.M. Influence of aging on the synthesis and morphology of the aggrecans synthesized by differentiated human articular chondrocytes. Osteoarthr. Cartil. 2000, 8, 170–179.
- Vinatier, C.; Dominguez, E.; Guicheux, J.; Carames, B. Role of the Inflammation-Autophagy-Senescence Integrative Network in Osteoarthritis. Front. Physiol. 2018, 9, 706.
- Herranz, N.; Gil, J. Mechanisms and functions of cellular senescence. J. Clin. Investig. 2018, 128, 1238–1246.
- Loeser, R.F. Aging and osteoarthritis: The role of chondrocyte senescence and aging changes in the cartilage matrix. Osteoarthr. Cartil. 2009, 17, 971–979.
- Liu, Y.; Zhang, Z.; Li, T.; Xu, H.; Zhang, H. Senescence in osteoarthritis: From mechanism to potential treatment. Arthritis Res. Ther. 2022, 24, 174.
- Jeon, O.H.; David, N.; Campisi, J.; Elisseeff, J.H. Senescent cells and osteoarthritis: A painful connection. J. Clin. Investig. 2018, 128, 1229–1237.
- Eckstein, F.; Hudelmaier, M.; Putz, R. The effects of exercise on human articular cartilage. J. Anat. 2006, 208, 491–512.
- Eskelinen, A.S.A.; Florea, C.; Tanska, P.; Hung, H.K.; Frank, E.H.; Mikkonen, S.; Nieminen, P.; Julkunen, P.; Grodzinsky, A.J.; Korhonen, R.K. Cyclic loading regime considered beneficial does not protect injured and interleukin-1-inflamed cartilage from post-traumatic osteoarthritis. J. Biomech. 2022, 141, 111181.
- Fu, S.; Thompson, C.L.; Ali, A.; Wang, W.; Chapple, J.P.; Mitchison, H.M.; Beales, P.L.; Wann, A.K.T.; Knight, M.M. Mechanical loading inhibits cartilage inflammatory signalling via an HDAC6 and IFT-dependent mechanism regulating primary cilia elongation. Osteoarthr. Cartil. 2019, 27, 1064–1074.
- Zheng, W.; Li, X.; Li, J.; Wang, X.; Liu, D.; Zhai, L.; Ding, B.; Li, G.; Sun, Y.; Yokota, H.; et al. Mechanical loading mitigates osteoarthritis symptoms by regulating the inflammatory microenvironment in a mouse model. Ann. N. Y. Acad. Sci. 2022, 1512, 141–153.
- Martin, J.A.; Anderson, D.D.; Goetz, J.E.; Fredericks, D.; Pedersen, D.R.; Ayati, B.P.; Marsh, J.L.; Buckwalter, J.A. Complementary models reveal cellular responses to contact stresses that contribute to post-traumatic osteoarthritis. J. Orthop. Res. Off. Publ. Orthop. Res. Soc. 2017, 35, 515–523.
- O’Conor, C.J.; Leddy, H.A.; Benefield, H.C.; Liedtke, W.B.; Guilak, F. TRPV4-mediated mechanotransduction regulates the metabolic response of chondrocytes to dynamic loading. Proc. Natl. Acad. Sci. USA 2014, 111, 1316–1321.
- O’Conor, C.J.; Ramalingam, S.; Zelenski, N.A.; Benefield, H.C.; Rigo, I.; Little, D.; Wu, C.L.; Chen, D.; Liedtke, W.; McNulty, A.L.; et al. Cartilage-Specific Knockout of the Mechanosensory Ion Channel TRPV4 Decreases Age-Related Osteoarthritis. Sci. Rep. 2016, 6, 29053.
- Lee, W.; Leddy, H.A.; Chen, Y.; Lee, S.H.; Zelenski, N.A.; McNulty, A.L.; Wu, J.; Beicker, K.N.; Coles, J.; Zauscher, S.; et al. Synergy between Piezo1 and Piezo2 channels confers high-strain mechanosensitivity to articular cartilage. Proc. Natl. Acad. Sci. USA 2014, 111, E5114–E5122.
- Bubolz, A.H.; Mendoza, S.A.; Zheng, X.; Zinkevich, N.S.; Li, R.; Gutterman, D.D.; Zhang, D.X. Activation of endothelial TRPV4 channels mediates flow-induced dilation in human coronary arterioles: Role of Ca2+ entry and mitochondrial ROS signaling. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H634–H642.
- Knight, M.M.; Bomzon, Z.; Kimmel, E.; Sharma, A.M.; Lee, D.A.; Bader, D.L. Chondrocyte Deformation Induces Mitochondrial Distortion and Heterogeneous Intracellular Strain Fields. Biomech. Model. Mechanobiol. 2006, 5, 180.
- Koike, M.; Nojiri, H.; Ozawa, Y.; Watanabe, K.; Muramatsu, Y.; Kaneko, H.; Morikawa, D.; Kobayashi, K.; Saita, Y.; Sasho, T.; et al. Mechanical overloading causes mitochondrial superoxide and SOD2 imbalance in chondrocytes resulting in cartilage degeneration. Sci. Rep. 2015, 5, 11722.
- Hirose, N.; Okamoto, Y.; Yanoshita, M.; Asakawa, Y.; Sumi, C.; Takano, M.; Nishiyama, S.; Su, S.C.; Mitsuyoshi, T.; Kunimatsu, R.; et al. Protective effects of cilengitide on inflammation in chondrocytes under excessive mechanical stress. Cell Biol. Int. 2020, 44, 966–974.
- Bevill, S.; Boyer, K.; Andriacchi, T. The regional sensitivity of chondrocyte gene expression to coactive mechanical load and exogenous TNF-alpha stimuli. J. Biomech. Eng. 2014, 136, 091005.
- Wang, T.; He, C. Pro-inflammatory cytokines: The link between obesity and osteoarthritis. Cytokine Growth Factor Rev. 2018, 44, 38–50.
- Vincent, T.L. Targeting mechanotransduction pathways in osteoarthritis: A focus on the pericellular matrix. Curr. Opin. Pharmacol. 2013, 13, 449–454.
- Tang, X.; Muhammad, H.; McLean, C.; Miotla-Zarebska, J.; Fleming, J.; Didangelos, A.; Önnerfjord, P.; Leask, A.; Saklatvala, J.; Vincent, T.L. Connective tissue growth factor contributes to joint homeostasis and osteoarthritis severity by controlling the matrix sequestration and activation of latent TGFβ. Ann. Rheum. Dis. 2018, 77, 1372–1380.
- Yang, X.; Chen, L.; Xu, X.; Li, C.; Huang, C.; Deng, C.X. TGF-beta/Smad3 signals repress chondrocyte hypertrophic differentiation and are required for maintaining articular cartilage. J. Cell Biol. 2001, 153, 35–46.
- Lires-Deán, M.; Caramés, B.; Cillero-Pastor, B.; Galdo, F.; López-Armada, M.J.; Blanco, F.J. Anti-apoptotic effect of transforming growth factor-beta1 on human articular chondrocytes: Role of protein phosphatase 2A. Osteoarthr. Cartil. 2008, 16, 1370–1378.
- Wang, G.; Chen, S.; Xie, Z.; Shen, S.; Xu, W.; Chen, W.; Li, X.; Wu, Y.; Li, L.; Liu, B.; et al. TGFβ attenuates cartilage extracellular matrix degradation via enhancing FBXO6-mediated MMP14 ubiquitination. Ann. Rheum. Dis. 2020, 79, 1111–1120.
- Madej, W.; van Caam, A.; Blaney Davidson, E.N.; van der Kraan, P.M.; Buma, P. Physiological and excessive mechanical compression of articular cartilage activates Smad2/3P signaling. Osteoarthr. Cartil. 2014, 22, 1018–1025.
- Zhen, G.; Wen, C.; Jia, X.; Li, Y.; Crane, J.L.; Mears, S.C.; Askin, F.B.; Frassica, F.J.; Chang, W.; Yao, J.; et al. Inhibition of TGF-β signaling in mesenchymal stem cells of subchondral bone attenuates osteoarthritis. Nat. Med. 2013, 19, 704–712.
- Zhen, G.; Guo, Q.; Li, Y.; Wu, C.; Zhu, S.; Wang, R.; Guo, X.E.; Kim, B.C.; Huang, J.; Hu, Y.; et al. Mechanical stress determines the configuration of TGFbeta activation in articular cartilage. Nat. Commun. 2021, 12, 1706.
- Chia, S.L.; Sawaji, Y.; Burleigh, A.; McLean, C.; Inglis, J.; Saklatvala, J.; Vincent, T. Fibroblast growth factor 2 is an intrinsic chondroprotective agent that suppresses ADAMTS-5 and delays cartilage degradation in murine osteoarthritis. Arthritis Rheum. 2009, 60, 2019–2027.
- Thomas, R.S.; Clarke, A.R.; Duance, V.C.; Blain, E.J. Effects of Wnt3A and mechanical load on cartilage chondrocyte homeostasis. Arthritis Res. Ther. 2011, 13, R203.
- De Palma, A.; Cheleschi, S.; Pascarelli, N.A.; Giannotti, S.; Galeazzi, M.; Fioravanti, A. Hydrostatic pressure as epigenetic modulator in chondrocyte cultures: A study on miRNA-155, miRNA-181a and miRNA-223 expression levels. J. Biomech. 2018, 66, 165–169.
- Choi, M.C.; Jo, J.; Park, J.; Kang, H.K.; Park, Y. NF-κB Signaling Pathways in Osteoarthritic Cartilage Destruction. Cells 2019, 8, 734.
- Chang, S.H.; Mori, D.; Kobayashi, H.; Mori, Y.; Nakamoto, H.; Okada, K.; Taniguchi, Y.; Sugita, S.; Yano, F.; Chung, U.I.; et al. Excessive mechanical loading promotes osteoarthritis through the gremlin-1-NF-kappaB pathway. Nat. Commun. 2019, 10, 1442.
- Devasagayam, T.P.; Tilak, J.C.; Boloor, K.K.; Sane, K.S.; Ghaskadbi, S.S.; Lele, R.D. Free radicals and antioxidants in human health: Current status and future prospects. J. Assoc. Physicians India 2004, 52, 794–804.
- Birben, E.; Sahiner, U.M.; Sackesen, C.; Erzurum, S.; Kalayci, O. Oxidative stress and antioxidant defense. World Allergy Organ. J. 2012, 5, 9–19.
- Blanco, F.J.; Rego, I.; Ruiz-Romero, C. The role of mitochondria in osteoarthritis. Nat. Rev. Rheumatol. 2011, 7, 161–169.
- Dai, Y.; Liu, S.; Li, J.; Li, J.; Lan, Y.; Nie, H.; Zuo, Y. SIRT4 suppresses the inflammatory response and oxidative stress in osteoarthritis. Am. J. Transl. Res. 2020, 12, 1965–1975.
- Altindag, O.; Erel, O.; Aksoy, N.; Selek, S.; Celik, H.; Karaoglanoglu, M. Increased oxidative stress and its relation with collagen metabolism in knee osteoarthritis. Rheumatol. Int. 2007, 27, 339–344.
- Adam-Vizi, V.; Chinopoulos, C. Bioenergetics and the formation of mitochondrial reactive oxygen species. Trends Pharmacol. Sci. 2006, 27, 639–645.
- Wegner, A.M.; Campos, N.R.; Robbins, M.A.; Haddad, A.F.; Cunningham, H.C.; Yik, J.H.N.; Christiansen, B.A.; Haudenschild, D.R. Acute Changes in NADPH Oxidase 4 in Early Post-Traumatic Osteoarthritis. J. Orthop. Res. Off. Publ. Orthop. Res. Soc. 2019, 37, 2429–2436.
- Hiran, T.S.; Moulton, P.J.; Hancock, J.T. Detection of superoxide and NADPH oxidase in porcine articular chondrocytes. Free Radic. Biol. Med. 1997, 23, 736–743.
- Förstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837.
- Mendes, A.F.; Carvalho, A.P.; Caramona, M.M.; Lopes, M.C. Role of nitric oxide in the activation of NF-kappaB, AP-1 and NOS II expression in articular chondrocytes. Inflamm. Res. 2002, 51, 369–375.
- van den Berg, W.B.; van de Loo, F.; Joosten, L.A.; Arntz, O.J. Animal models of arthritis in NOS2-deficient mice. Osteoarthr. Cartil. 1999, 7, 413–415.
- Ahmad, N.; Ansari, M.Y.; Bano, S.; Haqqi, T.M. Imperatorin suppresses IL-1beta-induced iNOS expression via inhibiting ERK-MAPK/AP1 signaling in primary human OA chondrocytes. Int. Immunopharmacol. 2020, 85, 106612.
- Fermor, B.; Weinberg, J.B.; Pisetsky, D.S.; Misukonis, M.A.; Banes, A.J.; Guilak, F. The effects of static and intermittent compression on nitric oxide production in articular cartilage explants. J. Orthop. Res. Off. Publ. Orthop. Res. Soc. 2001, 19, 729–737.
- Blanco, F.J.; Ochs, R.L.; Schwarz, H.; Lotz, M. Chondrocyte apoptosis induced by nitric oxide. Am. J. Pathol. 1995, 146, 75–85.
- Charlier, E.; Relic, B.; Deroyer, C.; Malaise, O.; Neuville, S.; Collée, J.; Malaise, M.G.; De Seny, D. Insights on Molecular Mechanisms of Chondrocytes Death in Osteoarthritis. Int. J. Mol. Sci. 2016, 17, 2146.
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950.
- Grishko, V.I.; Ho, R.; Wilson, G.L.; Pearsall, A.W.T. Diminished mitochondrial DNA integrity and repair capacity in OA chondrocytes. Osteoarthr. Cartil. 2009, 17, 107–113.
- Liu, L.; Luo, P.; Yang, M.; Wang, J.; Hou, W.; Xu, P. The role of oxidative stress in the development of knee osteoarthritis: A comprehensive research review. Front. Mol. Biosci. 2022, 9, 1001212.
- Johnson, K.; Jung, A.; Murphy, A.; Andreyev, A.; Dykens, J.; Terkeltaub, R. Mitochondrial oxidative phosphorylation is a downstream regulator of nitric oxide effects on chondrocyte matrix synthesis and mineralization. Arthritis Rheum. 2000, 43, 1560–1570.
- Collins, J.A.; Wood, S.T.; Nelson, K.J.; Rowe, M.A.; Carlson, C.S.; Chubinskaya, S.; Poole, L.B.; Furdui, C.M.; Loeser, R.F. Oxidative Stress Promotes Peroxiredoxin Hyperoxidation and Attenuates Pro-survival Signaling in Aging Chondrocytes. J. Biol. Chem. 2016, 291, 6641–6654.
- Kim, S.; Lee, K.; Choi, Y.S.; Ku, J.; Kim, H.; Kharbash, R.; Yoon, J.; Lee, Y.S.; Kim, J.H.; Lee, Y.J.; et al. Mitochondrial double-stranded RNAs govern the stress response in chondrocytes to promote osteoarthritis development. Cell Rep. 2022, 40, 111178.
- Yin, W.; Park, J.I.; Loeser, R.F. Oxidative stress inhibits insulin-like growth factor-I induction of chondrocyte proteoglycan synthesis through differential regulation of phosphatidylinositol 3-Kinase-Akt and MEK-ERK MAPK signaling pathways. J. Biol. Chem. 2009, 284, 31972–31981.
- Yasuhara, R.; Miyamoto, Y.; Akaike, T.; Akuta, T.; Nakamura, M.; Takami, M.; Morimura, N.; Yasu, K.; Kamijo, R. Interleukin-1beta induces death in chondrocyte-like ATDC5 cells through mitochondrial dysfunction and energy depletion in a reactive nitrogen and oxygen species-dependent manner. Biochem. J. 2005, 389, 315–323.
- Joos, H.; Wildner, A.; Hogrefe, C.; Reichel, H.; Brenner, R.E. Interleukin-1 beta and tumor necrosis factor alpha inhibit migration activity of chondrogenic progenitor cells from non-fibrillated osteoarthritic cartilage. Arthritis Res. Ther. 2013, 15, R119.
- Dizdaroglu, M.; Jaruga, P. Mechanisms of free radical-induced damage to DNA. Free. Radic. Res. 2012, 46, 382–419.
- Aivaliotis, I.L.; Pateras, I.S.; Papaioannou, M.; Glytsou, C.; Kontzoglou, K.; Johnson, E.O.; Zoumpourlis, V. How Do Cytokines Trigger Genomic Instability? J. Biomed. Biotechnol. 2012, 2012, 536761.
- Ou, H.L.; Schumacher, B. DNA damage responses and p53 in the aging process. Blood 2018, 131, 488–495.
- Copp, M.E.; Chubinskaya, S.; Bracey, D.N.; Shine, J.; Sessions, G.; Loeser, R.F.; Diekman, B.O. Comet assay for quantification of the increased DNA damage burden in primary human chondrocytes with aging and osteoarthritis. Aging Cell 2022, 21, e13698.
- Chen, A.F.; Davies, C.M.; De Lin, M.; Fermor, B. Oxidative DNA damage in osteoarthritic porcine articular cartilage. J. Cell. Physiol. 2008, 217, 828–833.
- Copp, M.E.; Flanders, M.C.; Gagliardi, R.; Gilbertie, J.M.; Sessions, G.A.; Chubinskaya, S.; Loeser, R.F.; Schnabel, L.V.; Diekman, B.O. The combination of mitogenic stimulation and DNA damage induces chondrocyte senescence. Osteoarthr. Cartil. 2021, 29, 402–412.
- Kim, J.; Xu, M.; Xo, R.; Mates, A.; Wilson, G.L.; Pearsall, A.W.t.; Grishko, V. Mitochondrial DNA damage is involved in apoptosis caused by pro-inflammatory cytokines in human OA chondrocytes. Osteoarthr. Cartil. 2010, 18, 424–432.
- Davies, C.M.; Guilak, F.; Weinberg, J.B.; Fermor, B. Reactive nitrogen and oxygen species in interleukin-1-mediated DNA damage associated with osteoarthritis. Osteoarthr. Cartil. 2008, 16, 624–630.
- Bang, J.; Kang, D.; Jung, J.; Yoo, T.J.; Shim, M.S.; Gladyshev, V.N.; Tsuji, P.A.; Hatfield, D.L.; Kim, J.H.; Lee, B.J. SEPHS1: Its evolution, function and roles in development and diseases. Arch. Biochem. Biophys. 2022, 730, 109426.
- Kang, D.; Lee, J.; Jung, J.; Carlson, B.A.; Chang, M.J.; Chang, C.B.; Kang, S.B.; Lee, B.C.; Gladyshev, V.N.; Hatfield, D.L.; et al. Selenophosphate synthetase 1 deficiency exacerbates osteoarthritis by dysregulating redox homeostasis. Nat. Commun. 2022, 13, 779.
- Takayama, K.; Kawakami, Y.; Lee, S.; Greco, N.; Lavasani, M.; Mifune, Y.; Cummins, J.H.; Yurube, T.; Kuroda, R.; Kurosaka, M.; et al. Involvement of ERCC1 in the pathogenesis of osteoarthritis through the modulation of apoptosis and cellular senescence. J. Orthop. Res. Off. Publ. Orthop. Res. Soc. 2014, 32, 1326–1332.
- Guo, Q.; Chen, X.; Chen, J.; Zheng, G.; Xie, C.; Wu, H.; Miao, Z.; Lin, Y.; Wang, X.; Gao, W.; et al. STING promotes senescence, apoptosis, and extracellular matrix degradation in osteoarthritis via the NF-κB signaling pathway. Cell Death Dis. 2021, 12, 13.
- Zhang, X.; Xiang, S.; Zhang, Y.; Liu, S.; Lei, G.; Hines, S.; Wang, N.; Lin, H. In vitro study to identify ligand-independent function of estrogen receptor-alpha in suppressing DNA damage-induced chondrocyte senescence. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2023, 37, e22746.
- Wang, N.; Zhang, X.; Rothrauff, B.B.; Fritch, M.R.; Chang, A.; He, Y.; Yeung, M.; Liu, S.; Lipa, K.E.; Lei, G.; et al. Novel role of estrogen receptor-alpha on regulating chondrocyte phenotype and response to mechanical loading. Osteoarthr. Cartil. 2022, 30, 302–314.
- Hipp, M.S.; Kasturi, P.; Hartl, F.U. The proteostasis network and its decline in ageing. Nat. Rev. Mol. Cell Biol. 2019, 20, 421–435.
- Rellmann, Y.; Eidhof, E.; Dreier, R. Review: ER stress-induced cell death in osteoarthritic cartilage. Cell Signal. 2021, 78, 109880.
- Kung, L.H.; Rajpar, M.H.; Briggs, M.D.; Boot-Handford, R.P. Hypertrophic chondrocytes have a limited capacity to cope with increases in endoplasmic reticulum stress without triggering the unfolded protein response. J. Histochem. Cytochem. Off. J. Histochem. Soc. 2012, 60, 734–748.
- Hetz, C.; Zhang, K.; Kaufman, R.J. Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell Biol. 2020, 21, 421–438.
- Piróg, K.A.; Jaka, O.; Katakura, Y.; Meadows, R.S.; Kadler, K.E.; Boot-Handford, R.P.; Briggs, M.D. A mouse model offers novel insights into the myopathy and tendinopathy often associated with pseudoachondroplasia and multiple epiphyseal dysplasia. Hum. Mol. Genet. 2010, 19, 52–64.
- Nugent, A.E.; Speicher, D.M.; Gradisar, I.; McBurney, D.L.; Baraga, A.; Doane, K.J.; Horton, W.E., Jr. Advanced osteoarthritis in humans is associated with altered collagen VI expression and upregulation of ER-stress markers Grp78 and bag-1. J. Histochem. Cytochem. Off. J. Histochem. Soc. 2009, 57, 923–931.
- Yang, L.; McBurney, D.; Tang, S.C.; Carlson, S.G.; Horton, W.E., Jr. A novel role for Bcl-2 associated-athanogene-1 (Bag-1) in regulation of the endoplasmic reticulum stress response in mammalian chondrocytes. J. Cell. Biochem. 2007, 102, 786–800.
- Hamamura, K.; Goldring, M.B.; Yokota, H. Involvement of p38 MAPK in regulation of MMP13 mRNA in chondrocytes in response to surviving stress to endoplasmic reticulum. Arch. Oral Biol. 2009, 54, 279–286.
- Takada, K.; Hirose, J.; Senba, K.; Yamabe, S.; Oike, Y.; Gotoh, T.; Mizuta, H. Enhanced apoptotic and reduced protective response in chondrocytes following endoplasmic reticulum stress in osteoarthritic cartilage. Int. J. Exp. Pathol. 2011, 92, 232–242.
- Tan, L.; Register, T.C.; Yammani, R.R. Age-Related Decline in Expression of Molecular Chaperones Induces Endoplasmic Reticulum Stress and Chondrocyte Apoptosis in Articular Cartilage. Aging Dis. 2020, 11, 1091–1102.
- Feng, Y.; Li, B.; Li, S.J.; Yang, X.C.; Lv, T.T.; Shang, H.; Wu, Z.B.; Zhang, Y. Skp2/p27 axis regulates chondrocyte proliferation under high glucose induced endoplasmic reticulum stress. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 9129–9138.
- Tan, L.; Harper, L.; McNulty, M.A.; Carlson, C.S.; Yammani, R.R. High-fat diet induces endoplasmic reticulum stress to promote chondrocyte apoptosis in mouse knee joints. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2020, 34, 5818–5826.
- Eymard, F.; Parsons, C.; Edwards, M.H.; Petit-Dop, F.; Reginster, J.Y.; Bruyère, O.; Richette, P.; Cooper, C.; Chevalier, X. Diabetes is a risk factor for knee osteoarthritis progression. Osteoarthr. Cartil. 2015, 23, 851–859.
- Xie, Y.; Zhou, W.; Zhong, Z.; Zhao, Z.; Yu, H.; Huang, Y.; Zhang, P. Metabolic syndrome, hypertension, and hyperglycemia were positively associated with knee osteoarthritis, while dyslipidemia showed no association with knee osteoarthritis. Clin. Rheumatol. 2021, 40, 711–724.
- Courties, A.; Gualillo, O.; Berenbaum, F.; Sellam, J. Metabolic stress-induced joint inflammation and osteoarthritis. Osteoarthr. Cartil. 2015, 23, 1955–1965.
- Gierman, L.M.; van der Ham, F.; Koudijs, A.; Wielinga, P.Y.; Kleemann, R.; Kooistra, T.; Stoop, R.; Kloppenburg, M.; van Osch, G.J.; Stojanovic-Susulic, V.; et al. Metabolic stress-induced inflammation plays a major role in the development of osteoarthritis in mice. Arthritis Rheum. 2012, 64, 1172–1181.
- Ratneswaran, A.; Sun, M.M.; Dupuis, H.; Sawyez, C.; Borradaile, N.; Beier, F. Nuclear receptors regulate lipid metabolism and oxidative stress markers in chondrocytes. J. Mol. Med. 2017, 95, 431–444.
- Alvarez-Garcia, O.; Rogers, N.H.; Smith, R.G.; Lotz, M.K. Palmitate has proapoptotic and proinflammatory effects on articular cartilage and synergizes with interleukin-1. Arthritis Rheumatol. 2014, 66, 1779–1788.
- Lee, S.W.; Rho, J.H.; Lee, S.Y.; Chung, W.T.; Oh, Y.J.; Kim, J.H.; Yoo, S.H.; Kwon, W.Y.; Bae, J.Y.; Seo, S.Y.; et al. Dietary fat-associated osteoarthritic chondrocytes gain resistance to lipotoxicity through PKCK2/STAMP2/FSP27. Bone Res. 2018, 6, 20.
- van Gastel, N.; Stegen, S.; Eelen, G.; Schoors, S.; Carlier, A.; Daniëls, V.W.; Baryawno, N.; Przybylski, D.; Depypere, M.; Stiers, P.J.; et al. Lipid availability determines fate of skeletal progenitor cells via SOX9. Nature 2020, 579, 111–117.
- Donovan, E.L.; Lopes, E.B.P.; Batushansky, A.; Kinter, M.; Griffin, T.M. Independent effects of dietary fat and sucrose content on chondrocyte metabolism and osteoarthritis pathology in mice. Dis. Model. Mech. 2018, 11, dmm034827.
- Lee, S.Y.; Abel, E.D.; Long, F. Glucose metabolism induced by Bmp signaling is essential for murine skeletal development. Nat. Commun. 2018, 9, 4831.
- Huang, J.; Zhao, L.; Chen, D. Growth factor signalling in osteoarthritis. Growth Factors 2018, 36, 187–195.
- Suzuki, A.; Yabu, A.; Nakamura, H. Advanced glycation end products in musculoskeletal system and disorders. Methods 2020, 203, 179–186.
- Chen, Y.J.; Chan, D.C.; Chiang, C.K.; Wang, C.C.; Yang, T.H.; Lan, K.C.; Chao, S.C.; Tsai, K.S.; Yang, R.S.; Liu, S.H. Advanced glycation end-products induced VEGF production and inflammatory responses in human synoviocytes via RAGE-NF-κB pathway activation. J. Orthop. Res. Off. Publ. Orthop. Res. Soc. 2016, 34, 791–800.
- Yamabe, S.; Hirose, J.; Uehara, Y.; Okada, T.; Okamoto, N.; Oka, K.; Taniwaki, T.; Mizuta, H. Intracellular accumulation of advanced glycation end products induces apoptosis via endoplasmic reticulum stress in chondrocytes. FEBS J. 2013, 280, 1617–1629.
More