Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Conner Chen and Version 1 by Maja Jazvinšćak Jembrek.
Major depressive disorder is a severe neuropsychiatric disease driven by various hereditary, environmental, and psychological factors. It disables normal functioning and quality of life.
- depression
- flavonols
- neuroinflammation
1. Introduction
Major depressive disorder is a severe neuropsychiatric disease driven by various hereditary, environmental, and psychological factors [1]. It disables normal functioning and quality of life. Typical symptoms include ongoing sadness, feelings of guilt, fatigue, loss of energy, lack of motivation for engaging in previously rewarding activities, impairment of cognitive functions, and disturbances of sleep, weight, and appetite. Depression is a common mental disorder and is one of the leading causes of disability worldwide. Globally, more than 350 million people suffer from depression, with the prevalence being higher among women [2,3][2][3].
The clinical manifestations of depression are accompanied by morphological changes of specific brain regions mainly related to mood regulation, rewards, emotions, and cognitive functions (such as decision making and memory). Neuroimaging studies have revealed significant structural alterations, together with abnormal interregional connectivity and brain activity, in various cortical and limbic brain areas. Affected areas include the frontal lobe (e.g., the orbitofrontal and dorsolateral prefrontal cortexes), the hippocampus, the thalamus, the basal ganglia, the amygdala, and the anterior cingulate cortex as well as cerebellar neurons projecting toward the ventral tegmental area (VTA) [4,5,6,7,8,9][4][5][6][7][8][9]. However, due to the diversity of demographic and clinical data, methodologies used, and study designs, the results concerning the identification of the depression-related patterns of brain changes are very heterogeneous and far from generalization.
2. Neuropathological Mechanisms Underlying the Development of Depression
2.1. Monoamine Hypothesis of Depression
The mechanisms underlying the etiology of depression are still not fully understood. From a clinical perspective, the typical abnormality witnessed is a disruption of neurotransmitter levels, particularly those of serotonin, dopamine, and norepinephrine. Depressed patients have reduced levels of monoamines and their metabolites in the cerebrospinal fluid and the post-mortem brain, although the results on this matter are not completely uniform and conclusive [10,11,12,13][10][11][12][13]. The major mechanisms related to monoamine depletion include the impaired activity of enzymes participating in their degradation, the impaired activity of enzymes involved in tryptophan synthesis, and the impaired activity of monoamine transporters and receptors [14,15,16][14][15][16]. Moreover, most of the behavioral symptoms of depressed patients can be related to disruptions in monoamine levels [17]. Hence, the monoamine hypothesis contends that monoamine deficiency is the core mechanism underlying the pathophysiological events of depression [18]. This hypothesis is supported by the mechanisms of action of clinically used antidepressants. The main pharmacological approaches are based on improving the monoamine balance by using selective serotonin reuptake inhibitors (SSRIs); tricyclic antidepressants that inhibit the reuptake of serotonin, dopamine, and norepinephrine; monoamine oxidase inhibitors; and selective norepinephrine reuptake inhibitors (NARI) [19,20][19][20]. The first-line drugs employed are usually SSPIs such as fluoxetine, sertraline, and citalopram, which slowly increase synaptic serotonin levels. However, despite this repertoire of available drugs, many patients do not respond adequately to such therapy [21], and many unwanted side effects are induced alongside the beneficial ones. Some of the adverse effects reported by patients include dizziness, sedation, anxiety, cardiac problems, gastrointestinal and sexual disfunction, sleep disturbances, appetite changes, forgetfulness, confusion, difficulty finding words, and other cognitive impairments [22].
Besides the limited efficacy and slow onset of the most-prescribed drugs, monoamine depletion neither causes depression in healthy individuals nor worsens symptoms in patients not undergoing therapy, thereby raising questions concerning the exact role of monoamine deficiency in the etiopathology of depression and suggesting the involvement of other pathological mechanisms [18,23][18][23]. More recent studies have demonstrated neurochemical, structural, and functional impairments of GABAergic and glutamatergic systems. As will be explained below, these dysfunctions are likely mediated by excitotoxicity and increased levels of pro-inflammatory cytokines and adrenal glucocorticoids in combination with certain environmental factors [24]. Evidence revealing that the impaired function of GABAergic and glutamatergic systems is implicated in the pathophysiology of depression resulted in advancements in therapeutic strategies, particularly with respect to the treatment-resistant depression, leading to the introduction of fast-acting antidepressants targeting N-methyl-D-aspartate (NMDA) and γ-aminobutyric acid type A (GABAA) receptors [24,25,26][24][25][26].
2.2. Hippocampus and Depression
Brain imaging has revealed lesions mainly localized in the prefrontal and cingulate cortex, hippocampus, and amygdala [8[8][27],27], indicating increased susceptibility to neuronal death in distinct brain areas. Structural and functional alterations of the hippocampus are particularly relevant considering this structure’s role in the regulation of stress response, memory-related consolidation processes, and adult neurogenesis. Depressed patients have reduced hippocampal volume due to a decreased number of both neuronal and glial cells, suggesting that hippocampal changes are likely a contributing factor in the pathophysiology of depression [4,28,29][4][28][29]. At the cellular and molecular level, it is believed that the dysregulation of the HPA axis and the consequent increase in the quantity of glucocorticoids are the underlying mechanisms of reduced neurogenesis, excitotoxicity, inflammation, and depleted levels of neurotrophins, which all contribute to reducing the size of the hippocampus [4].
The severity of hippocampal degeneration positively correlates with the duration of the symptoms and negatively correlates with the efficacy of antidepressant therapies and clinical outcomes [30,31,32][30][31][32]. Boku et al. proposed two hypotheses for explaining changes in the hippocampal volume [33]. The neuroplasticity hypothesis emphasizes the degenerative changes of mature neurons in the hippocampus (the reduced branching and length of dendrites and the reduced density of dendritic spines), whereas the neurogenesis hypothesis considers the reduced neurogenesis and the role of the neural progenitor cells in the dentate gyrus. These hippocampal deteriorations are considered to be largely responsible for delayed responses to antidepressant therapy. It has been shown that only long-term treatment with antidepressants protects mature neurons and promotes the proliferation of neural progenitors and neurogenesis. At least partially, the protection of mature neurons is mediated through increased 5-HT4-receptor-mediated signaling [34], whereas neurogenesis is stimulated by glucocorticoid-receptor-initiated pathways [35]. Thus, the time needed to observe effects on hippocampal neurons corresponds with the therapeutic window required to observe the improvement of clinical manifestations [28,29][28][29].
Adult Neurogenesis and Depression
The process of neurogenesis is tightly related to neurotrophic factors. Brain-derived neurotrophic factor (BDNF) belongs to the neurotrophin family and is one of the most abundant neurotrophins in the brain. BDNF acts via the tropomyosin receptor kinase B (TrkB) receptor, which is expressed on both neuros and glia. The activation of TrkB receptors triggers the activation of signaling cascades involved in neuronal survival and functioning, including the mitogen-activated protein kinase (MAPK), phosphatidylinositol 3-kinase (PI3K)/Akt, and phospholipase C-γ (PLC-γ) pathways [36,37][36][37]. BDNF plays an important role in the wide spectrum of neurophysiological processes. Among other functions, it regulates neuronal development and survival, synaptogenesis, plasticity, and learning and memory [38,39,40,41][38][39][40][41]. Different BDNF roles are mediated by distinct transcripts that arise from the nine promotors in the BDNF gene, which all encode the same BDNF protein [42]. These transcripts are expressed in a complex spatio-temporal pattern, respond to different stimuli, and trigger the activation of distinct signaling networks [43,44][43][44]. Hence, depending on the brain region, the effects of BDNF could be antidepressant or pro-depressant. In the hippocampus and prefrontal cortex, BDNF displays antidepressant activity, and depressed patients usually exhibit reduced serum BDNF levels and levels of BDNF IV transcript in the hippocampus and prefrontal cortex [45,46,47][45][46][47]. The disruption of murine promotor IV impairs GABAergic transmission and the expression of monoaminergic genes in the hippocampus and prefrontal cortex and results in depression-like behavior [48,49,50][48][49][50]. Moreover, there are indications that the methylation of promotor IV and plasma BDNF levels can be used as a potential predictor of treatment response [51]. It has been proposed that serum BDNF concentrations may serve as the peripheral biomarker of the disease [45], yet its levels in the serum do not always correlate with the severity of symptoms [52,53,54][52][53][54]. However, BDNF concentrations increase after antidepressant therapy, and there is a good correlation between BDNF changes and improvements in depression scores [55,56][55][56].
2.3. HPA Axis and Depression
HPA hyperactivity is commonly observed in patients with chronic stress and depression [57,58,59][57][58][59]. The hippocampus possesses many receptors for glucocorticoids. Through a negative feedback mechanism, it regulates glucocorticoid release from the adrenal cortex, thus playing a critical role in the tuning of the HPA axis [60,61][60][61]. The HPA axis starts with the neurosecretory cells of the hypothalamus that release corticotrophin-releasing hormone (CRH). CRH stimulates the release of the adrenocorticotropic hormone (ACTH) from the anterior pituitary, whereas ACTH further stimulates the adrenal gland so that it both produces and releases glucocorticoids. In turn, through inhibitory feedback, the released glucocorticoids attenuate the production of CRH and ACTH via glucocorticoid and mineralocorticoid receptors. These receptors are particularly abundant in the hippocampus. The excitatory output from hippocampal neurons along with the activity of inhibitory GABAergic cells regulate CRF-releasing neurons in the hypothalamus. Under stressful conditions, the HPA axis escapes this regulatory mechanism and produces large amounts of glucocorticoids [60,62][60][62]. A long-term increase in glucocorticoid levels reduces dendritic branching and dendritic length and induces the death of mature neurons and progenitor cells, resulting in reduced hippocampal volume. Thus, the disruption of neuroendocrine regulation ultimately impairs the excitability, functions, and integrity of the hippocampus; reduces plasticity and neurogenesis; and promotes susceptibility to the development of depression [58,63,64,65,66,67,68,69][58][63][64][65][66][67][68][69].
2.4. Oxidative Stress and Depression
Depression is accompanied by increased oxidative stress and reduced concentrations of antioxidants in the plasma [70,71][70][71].
Briefly, oxidative stress refers to a condition in which there is an imbalance between the generation of reactive oxygen and nitrogen species (ROS/RNS) and the ability of various enzymatic and non-enzymatic mechanisms of endogenous defense to maintain their levels in a physiological range. In appropriate concentrations, ROS have important functions, acting as signaling molecules in various redox-sensitive signaling pathways. However, if present in excess, they disturb neuronal signaling and react with cellular lipids, proteins, and nucleic acids, thus threatening their structure and proper neuronal functioning [72,73,74,75][72][73][74][75]. The brain is particularly vulnerable to oxidative injury due to its high metabolic activity, high content of redox-active transition metals that act as initiators of ROS generation via Fenton chemistry, high content of poly-unsaturated fatty acids (PUFAs) that are highly prone to lipid peroxidation, and limited mechanisms of antioxidative defense, among other reasons [76].
More importantly, specific molecules released or exposed during oxidative injury may elicit an innate immune response in the brain (acting as danger-associated molecular patterns (DAMPS)) and trigger sterile inflammation [77,78][77][78]. Thus, oxidative stress contributes to the inflammatory response and the increased production of proinflammatory cytokines, which inevitably results in the appearance of depressive behavior (Figure 1). Hence, both oxidative stress and neuroinflammation have been recognized as underlying factors in the development of depression [77,79,80,81][77][79][80][81].
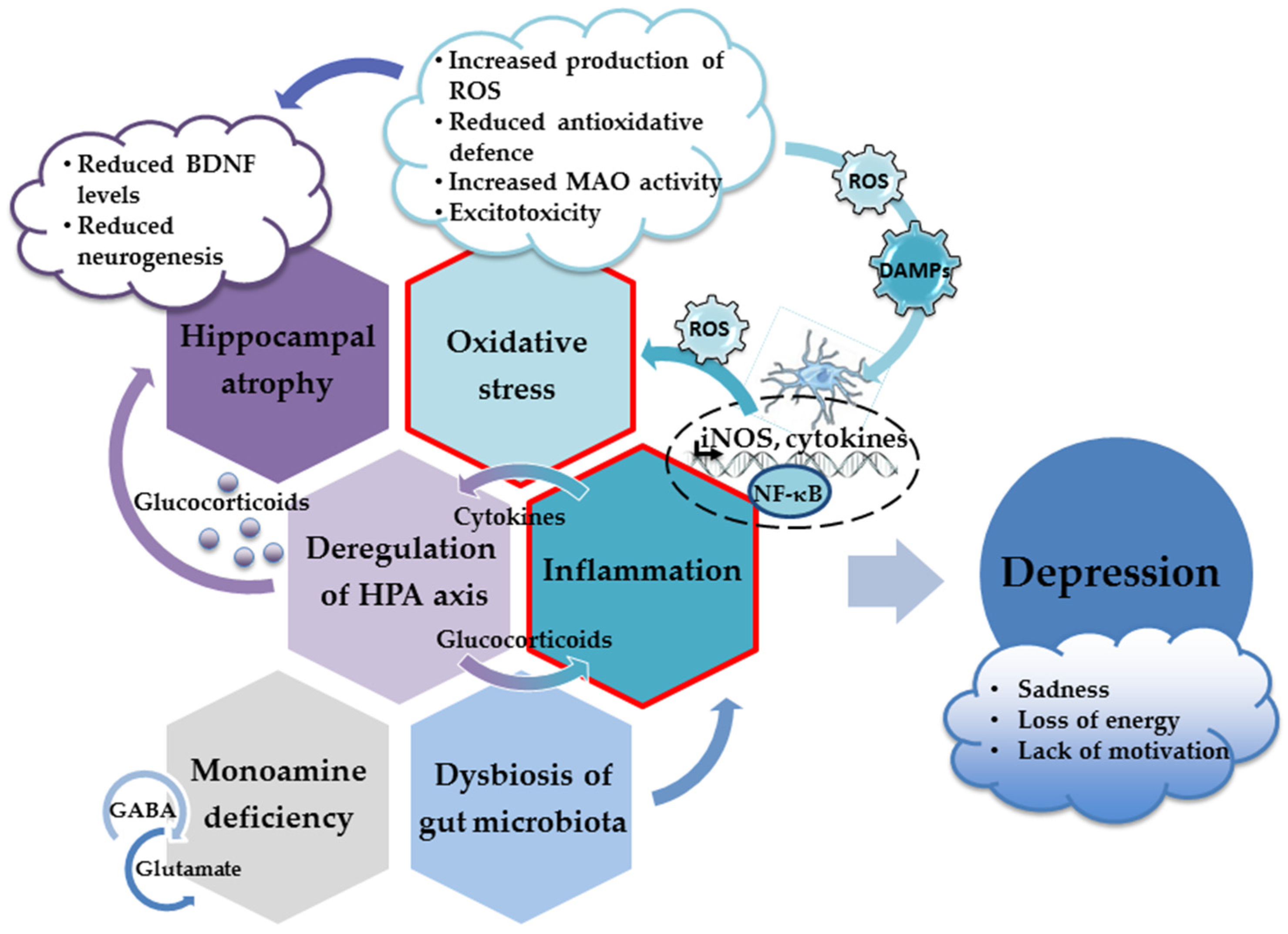
Figure 1.
The main neuropathological mechanisms involved in the development of depression.
The presence of oxidative stress markers is a common finding in depressed patients and animal models of depression [82]. 8-hydroxy-2′-deoxyguanosine (8-OHdG) is a typical indicator of oxidative DNA damage and it levels are significantly increased in blood samples from depressed patients [83,84][83][84]. The extent of lipid peroxidation in the peripheral blood is also increased and correlates with the severity of depressive symptoms [85,86,87,88][85][86][87][88]. Malondialdehyde (MDA), an end marker of lipid peroxidation, is usually found in increased concentrations in depressed patients [89]. Moreover, it has been suggested that certain oxidative stress indicators may be used as prognostic markers of disease severity and for the evaluation of the efficacy of administered antidepressants [82].
An increase in oxidative stress parameters reduces the content of intracellular mechanisms of antioxidative defense and jeopardizes neuronal protection. Thus, changes in the expression and activity of antioxidative and prooxidative enzymes have been observed; however, the results are not straightforward. Levels of the prooxidative enzyme xanthine oxidase, which generates superoxide anions and hydrogen peroxide, are usually increased in both depressed patients and animal models of depression [90,91][90][91]. On the other hand, levels of superoxide dismutase (SOD), an enzyme that decomposes superoxide to oxygen and hydrogen peroxide, were found to be reduced, unchanged, or enhanced in depressed patients [92,93,94,95][92][93][94][95]. Hydrogen peroxide is removed by catalase. Similarly, increased, unchanged, and decreased catalase activity has been reported [89,93,95][89][93][95]. Glutathione peroxidase also reduces hydrogen peroxide and regenerates reduced glutathione (GSH) from its oxidized pool (GSSG), thereby increasing the overall scavenging ability of GSH. The activity of glutathione peroxidase is predominantly found to be reduced in depressed patients, which is negatively correlated with the severity of the symptoms [93,96][93][96]. However, some studies did not observe changes in the glutathione peroxidase activity of depressed patients [84,92][84][92]. In yet another study, increases in the levels of glutathione peroxidase and SOD were dependent on the sample analyzed (plasma or erythrocytes) and clinical manifestations (depression with or without melancholia), suggesting that various factors affect the extent of changes in the activity of antioxidant enzymes in depressed patients [97]. Of note, the polymorphism of several genes participating in ROS metabolism may represent a risk for disease development and progression. It has been shown that single-nucleotide polymorphisms (SNPs) in genes encoding mitochondrial SOD (SOD2), catalase, and glutathione peroxidase 4 may modulate the risk of the onset of disease [98,99][98][99].
Quantities of non-enzymatic antioxidants are also disturbed. Besides developing oxidative stress that decreases the levels of these antioxidant molecules, depressed people often change their food preferences, which may deplete concentrations of specific dietary products, including vitamins [100]. Levels of vitamins A, C, and E as well as other small antioxidants (such as uric acid, albumin, and coenzyme Q10) are lower in depressed patients and negatively correlate with the disease’s severity [86,101,102,103][86][101][102][103]. Hence, various dietary approaches, such as supplementation with the bioactive antioxidative ingredients from certain types of food, have been considered as adjuvant strategies for mitigating depressive symptoms [104,105,106][104][105][106]. Several studies have shown that supplementation with vitamin C may induce antidepressant effects and improve moods [107,108][107][108]. However, in combination with citalopram therapy, vitamin C was as effective as a placebo [109], whereas in combination with fluoxetine, it significantly mitigated depressive symptoms in a pediatric population [110]. Similarly, results on supplementation with vitamin E are inconclusive and further studies are needed to clarify the possible benefits of vitamin E with respect to the management of depression [102,111,112][102][111][112]. Administration of N-acetyl cysteine, a GSH precursor, also provided promising evidence regarding its use as an adjuvant therapy for depression. Several studies have indicated its ability to reduce the severity of depression, improve moods, and increase the efficacy of standard antidepressant therapy, but these findings need further confirmation [113,114,115][113][114][115].
Metabolic reactions in the mitochondria are the major sources of ROS, but mitochondria are also the major targets of ROS action. Considering the principal role of mitochondria in determining cell death and survival via ATP production and the initiation of apoptosis, the preservation of mitochondrial functions is an important prerequisite for neuronal health [116,117][116][117]. In depressed patients, the expression and activity of enzymes participating in the mitochondrial respiratory chain are disrupted, leading to a reduction in ATP production [118,119][118][119]. A growing body of evidence is indicating that depression is related to mitochondrial dysfunction, which, consequently, disturbs ROS balance and promotes oxidative stress [120,121][120][121]. Depressed patients also have increased levels of circulating cell-free mtDNA (ccf-mtDNA), which likely reflects a fraction of the mitochondrial DNA released under cellular stress conditions [122].
Monoamine oxidase MAO-A and MAO-B, isoenzymes located in the outer mitochondrial membrane, catalyze the oxidative deamination of monoamines and deplete their levels in the brain, while also contributing to ROS production and oxidative stress. The MAO-A isoform is mainly involved in the metabolism of serotonin, dopamine, and norepinephrine, whereas MAO-B predominantly metabolizes dopamine. Increased activity of MAO-A is a common finding in depressed patients. It has been suggested that this upregulation is the predominant mechanism underlying monoamine loss [123,124,125][123][124][125]. Although MAO inhibitors were the first class of antidepressants developed and were used therapeutically for decades, based on concerns related to safety, possible side effects related to potential drug interactions, and dietary restrictions, they have been replaced with safer and more tolerable options. However, inhibitors of MAO activity are still considered the most valuable pharmacological option, particularly with respect to patients who fail to respond to first-line therapy [126,127,128][126][127][128].
Oxidative stress is usually accompanied by excitotoxicity, which is induced by the hyperactivation of glutamatergic NMDA receptors. It has been proposed that the impaired clearance and increased release of glutamate by activated glial cells upregulates glutamate levels and disturbs signaling through ionotropic and metabotropic glutamate receptors, thus contributing to neuronal dysfunction and, ultimately, behavioral changes [129]. Excitotoxicity also increases the production of NO, which, together with the superoxide anion, generates extremely cytotoxic peroxynitrite [130,131][130][131]. In addition, it has been shown that peroxynitrite inactivates tryptophan hydroxylase, the rate-limiting enzyme in serotonin synthesis [132]. Moreover, NO reacts with amino acid residues in proteins (mainly cysteine, tyrosine, tryptophan, and arginine) and causes protein oxidation, nitration, and nitrosylation, further threatening neuronal functioning [78,103,133][78][103][133]. However, peripheral measurements of nitrosative stress markers have provided variable results. For example, reduced levels of nitrate, without changes in NO and nitrite levels, have been reported in depressed patients [134].
There is yet another connection between depression and the activation of NMDA receptors. In inflammatory conditions, the kynurenine pathway and the production of kynurenine from tryptophan are potentiated, which per se reduce serotonin concentrations. On the other hand, kynurenine is metabolized by microglial enzymes, resulting in the production of various metabolites, some of which are neurotoxic, such as quinolinic acid, an NMDA receptor agonist [135,136][135][136].
Finally, the reduced expression of BDNF precipitates higher susceptibility to stress-induced oxidative damage, indicating an interplay between these depression-related parameters [137]. In turn, increased oxidative stress affects BDNF production and contributes to reduced neurogenesis and the dysregulation of hippocampal functioning [82].
2.5. Neuroinflammation, Cytokines, and Depression
Animal studies have provided evidence for a neuroinflammatory basis of depression, which entails sterile inflammation, glial activation, and the release of pro-inflammatory cytokines [138]. Depression is accompanied by a chronic, low-grade inflammatory state; the dysregulation of the innate and adaptive immune system; enhanced production of various cytokines, such as interleukin (IL)-1, IL-2, IL-6, IL-10, IL-12, IL-13, and IL-1β and tumor necrosis factor (TNF)-α; and decreased concentrations of interferon (INF)-γ [139,140,141,142,143][139][140][141][142][143]. Treatment with antidepressants reduces the severity of inflammation, while severe inflammation correlates with lower treatment efficacy [144,145][144][145].
The excessive production of cytokines, the signaling molecules of the immune system, affects neurocircuitry in the basal ganglia and anterior cingulate cortex, the functioning of the HPA axis, synaptic plasticity, and neurotransmission, leading to behavioral alterations, mood changes, and the impairment of cognitive functions characteristic of depression. The behavioral effects of cytokines are largely mediated through the activation of the inflammatory signaling pathways that regulate the production of monoamines, glutamate, neuropeptides, and BDNF [146,147,148,149][146][147][148][149].
An increase in glucocorticoid concentrations mediates the effects of cytokines on the activation of the HPA axis and the dysregulation of the inhibitory feedback mechanism. Thus, the inflammatory response stimulates the HPA axis directly (through cytokine action) and indirectly (through glucocorticoid resistance) [150]. In major depressive disorders with melancholic features, a positive correlation has been found between the severity of the disease and concentrations of cortisol and IL-6 [151]. On the other hand, there is evidence that antidepressants normalize levels of proinflammatory cytokines and restore the feedback inhibition of the HPA axis [145,152][145][152]. In addition, an increase in proinflammatory cytokine concentrations may induce the tryptophan–kynurenine pathway and the generation of neurotoxic end products, as previously explained [150].
Oxidative stress and uncontrolled production of ROS play important roles in the hyperactivation of the immune pathways and the upregulation of cytokine production [77,153][77][153]. Enhanced production of proinflammatory cytokines and oxidative stress are tightly intertwined processes. Excessive production of ROS results in the oxidative damage of various cellular structures and the production of danger signals (DAMPs) that initiate inflammatory responses and microglial activation. Following activation, microglia, the resident cells of the innate immune system in the brain, produce proinflammatory cytokines and ROS. In a vicious loop, the ROS produced induce oxidative stress, supporting the ongoing microglial activation and further increasing ROS concentrations [133,154,155][133][154][155].
DAMPs-initiated signaling cascades transduce the signal to the nucleus and activate transcription factor nuclear factor κB (NF-κB). The activation of the NF-κB cascade triggers the production of proinflammatory cytokines, reactive oxygen and nitrogen species, and other potentially neurotoxic molecules such as inducible nitric oxide synthase (iNOS). Increased activation of iNOS results in increased production of NO, which, in excess, acts as a powerful oxidant. As mentioned previously, the superoxide anion along with the NO produced by iNOS form extremely dangerous peroxynitrite radicals. Thus, sustained oxidative injury, the neuroinflammatory response, and microglial activation are largely driven by the NF-κB pathway, whereas this pathway’s inhibition prevents the induction of the inhibitory effects of chronic stress on neurogenesis and the appearance of depressive behavior [156,157][156][157]. In animal models of neuroinflammation and depression, the upregulation of the NF-κB pathway has been demonstrated in the prefrontal cortex and the hippocampus [158,159][158][159]. However, in the human population, the results are not so consistent. For example, the activation of the NF-κB pathway was not observed in the peripheral blood mononuclear cells of adolescents and depressed medical students [160,161][160][161].
Several enzymes that are downstream targets of NF-κB may affect the course of depression. Such enzymes include cyclooxygenase 2 (COX-2) and NADPH oxidase. COX-2 inhibitors have been observed to have a positive effect on neuronal inflammation and the severity of the depression [162[162][163][164],163,164], whereas a single-nucleotide polymorphism of the COX-2 gene may represent a risk for recurrent depressive disorder [165]. NADPH oxidase contributes to oxidative stress and neuroinflammation by overproducing the superoxide anion. As with COX-2, the pharmacological inhibition of NADPH oxidase presented beneficial antidepressant effects [166].
References
- Otte, C.; Gold, S.M.; Penninx, B.W.; Pariante, C.M.; Etkin, A.; Fava, M.; Mohr, D.C.; Schatzberg, A.F. Major depressive disorder. Nat. Rev. Dis. Prim. 2016, 2, 16065.
- Christensen, M.C.; Wong, C.M.J.; Baune, B.T. Symptoms of Major Depressive Disorder and Their Impact on Psychosocial Functioning in the Different Phases of the Disease: Do the Perspectives of Patients and Healthcare Providers Differ? Front. Psychiatry 2020, 11, 280.
- Lim, G.Y.; Tam, W.W.; Lu, Y.; Ho, C.S.; Zhang, M.W.; Ho, R.C. Prevalence of Depression in the Community from 30 Countries between 1994 and 2014. Sci. Rep. 2018, 8, 2861.
- Campbell, S.; Macqueen, G. The role of the hippocampus in the pathophysiology of major depression. J. Psychiatry Neurosci. 2004, 29, 417–426.
- Palmer, S.M.; Crewther, S.G.; Carey, L.M. START Project Team A meta-analysis of changes in brain activity in clinical depression. Front. Hum. Neurosci. 2015, 8, 1045.
- Zhang, F.F.; Peng, W.; Sweeney, J.A.; Jia, Z.Y.; Gong, Q.Y. Brain structure alterations in depression: Psychoradiological evidence. CNS Neurosci. Ther. 2018, 24, 994–1003.
- Helm, K.; Viol, K.; Weiger, T.M.; Tass, P.A.; Grefkes, C.; Del Monte, D.; Schiepek, G. Neuronal connectivity in major depressive disorder: A systematic review. Neuropsychiatr. Dis. Treat. 2018, 14, 2715–2737.
- Sindermann, L.; Redlich, R.; Opel, N.; Böhnlein, J.; Dannlowski, U.; Leehr, E.J. Systematic transdiagnostic review of magnetic-resonance imaging results: Depression, anxiety disorders and their co-occurrence. J. Psychiatr. Res. 2021, 142, 226–239.
- Baek, S.J.; Park, J.S.; Kim, J.; Yamamoto, Y.; Tanaka-Yamamoto, K. VTA-projecting cerebellar neurons mediate stress-dependent depression-like behaviors. eLife 2022, 11, e72981.
- Vermeiren, Y.; Van Dam, D.; Aerts, T.; Engelborghs, S.; De Deyn, P.P. Monoaminergic neurotransmitter alterations in postmortem brain regions of depressed and aggressive patients with Alzheimer’s disease. Neurobiol. Aging 2014, 35, 2691–2700.
- Kunugi, H.; Hori, H.; Ogawa, S. Biochemical markers subtyping major depressive disorder. Psychiatry Clin. Neurosci. 2015, 69, 597–608.
- Ogawa, S.; Tsuchimine, S.; Kunugi, H. Cerebrospinal fluid monoamine metabolite concentrations in depressive disorder: A meta-analysis of historic evidence. J. Psychiatr. Res. 2018, 105, 137–146.
- Silić, A.; Vukojević, J.; Peitl, V.; De Hert, M.; Karlović, D. Major depressive disorder: A possible typisation according to serotonin, inflammation, and metabolic syndrome. Acta Neuropsychiaty 2022, 34, 15–23.
- Shao, X.; Zhu, G. Associations Among Monoamine Neurotransmitter Pathways, Personality Traits, and Major Depressive Disorder. Front. Psychiatry 2020, 11, 381.
- Jacobsen, J.P.; Medvedev, I.O.; Caron, M.G. The 5-HT deficiency theory of depression: Perspectives from a naturalistic 5-HT deficiency model, the tryptophan hydroxylase 2Arg439His knockin mouse. Philos. Trans R. Soc. Lond. B Biol. Sci. 2012, 367, 2444–2459.
- Correia, A.S.; Vale, N. Tryptophan Metabolism in Depression: A Narrative Review with a Focus on Serotonin and Kynurenine Pathways. Int. J. Mol. Sci. 2022, 23, 8493.
- Jiang, Y.; Zou, D.; Li, Y.; Gu, S.; Dong, J.; Ma, X.; Xu, S.; Wang, F.; Huang, J.H. Monoamine Neurotransmitters Control Basic Emotions and Affect Major Depressive Disorders. Pharmaceuticals 2022, 15, 1203.
- Delgado, P.L. Depression: The case for a monoamine deficiency. J. Clin. Psychiatry 2000, 61 (Suppl. S6), 7–11.
- Gabriel, F.C.; de Melo, D.O.; Fráguas, R.; Leite-Santos, N.C.; Mantovani da Silva, R.A.; Ribeiro, E. Pharmacological treatment of depression: A systematic review comparing clinical practice guideline recommendations. PLoS ONE 2020, 15, e0231700.
- Elias, E.; Zhang, A.Y.; Manners, M.T. Novel Pharmacological Approaches to the Treatment of Depression. Life 2022, 12, 196.
- Kessler, R.C.; Berglund, P.; Demler, O.; Jin, R.; Koretz, D.; Merikangas, K.R.; Rush, A.J.; Walters, E.E.; Wang, P.S. National Comorbidity Survey Replication The epidemiology of major depressive disorder: Results from the National Comorbidity Survey Replication (NCS-R). JAMA 2003, 289, 3095–3105.
- Braund, T.A.; Tillman, G.; Palmer, D.M.; Gordon, E.; Rush, A.J.; Harris, A.W.F. Antidepressant side effects and their impact on treatment outcome in people with major depressive disorder: An iSPOT-D report. Transl. Psychiatry 2021, 11, 417.
- Li, Y.F. A hypothesis of monoamine (5-HT)—Glutamate/GABA long neural circuit: Aiming for fast-onset antidepressant discovery. Pharmacol. Ther. 2020, 208, 107494.
- Duman, R.S.; Sanacora, G.; Krystal, J.H. Altered Connectivity in Depression: GABA and Glutamate Neurotransmitter Deficits and Reversal by Novel Treatments. Neuron 2019, 102, 75–90.
- Taillefer de Laportalière, T.; Yrondi, A.; Jullien, A.; Cestac, P.; Montastruc, F. How to deprescribe esketamine in resistant depression? A point of view after first clinical uses. Epidemiol. Psychiatr. Sci. 2022, 31, e4.
- Kantrowitz, J.T.; Dong, Z.; Milak, M.S.; Rashid, R.; Kegeles, L.S.; Javitt, D.C.; Lieberman, J.A.; John Mann, J. Ventromedial prefrontal cortex/anterior cingulate cortex Glx, glutamate, and GABA levels in medication-free major depressive disorder. Transl. Psychiatry 2021, 11, 419.
- Pandya, M.; Altinay, M.; Malone, D.A., Jr.; Anand, A. Where in the brain is depression? Curr. Psychiatry Rep. 2012, 14, 634–642.
- Malberg, J.E.; Schechter, L.E. Increasing hippocampal neurogenesis: A novel mechanism for antidepressant drugs. Curr. Pharm. Des. 2005, 11, 145–155.
- Malykhin, N.V.; Carter, R.; Seres, P.; Coupland, N.J. Structural changes in the hippocampus in major depressive disorder: Contributions of disease and treatment. J. Psychiatry Neurosci. 2010, 35, 337–343.
- MacQueen, G.; Frodl, T. The hippocampus in major depression: Evidence for the convergence of the bench and bedside in psychiatric research? Mol. Psychiatry 2011, 16, 252–264.
- Roddy, D.W.; Farrell, C.; Doolin, K.; Roman, E.; Tozzi, L.; Frodl, T.; O’Keane, V.; O’Hanlon, E. The Hippocampus in Depression: More Than the Sum of Its Parts? Advanced Hippocampal Substructure Segmentation in Depression. Biol. Psychiatry 2019, 85, 487–497.
- Hsieh, M.H.; McQuoid, D.R.; Levy, R.M.; Payne, M.E.; MacFall, J.R.; Steffens, D.C. Hippocampal volume and antidepressant response in geriatric depression. Int. J. Geriatr. Psychiatry 2002, 17, 519–525.
- Boku, S.; Nakagawa, S.; Toda, H.; Hishimoto, A. Neural basis of major depressive disorder: Beyond monoamine hypothesis. Psychiatry Clin. Neurosci. 2018, 72, 3–12.
- Kobayashi, K.; Ikeda, Y.; Sakai, A.; Yamasaki, N.; Haneda, E.; Miyakawa, T.; Suzuki, H. Reversal of hippocampal neuronal maturation by serotonergic antidepressants. Proc. Natl. Acad. Sci. USA 2010, 107, 8434–8439.
- Anacker, C.; Zunszain, P.A.; Cattaneo, A.; Carvalho, L.A.; Garabedian, M.J.; Thuret, S.; Price, J.; Pariante, C.M. Antidepressants increase human hippocampal neurogenesis by activating the glucocorticoid receptor. Mol. Psychiatry 2011, 16, 738–750.
- Foltran, R.B.; Diaz, S.L. BDNF isoforms: A round trip ticket between neurogenesis and serotonin? J. Neurochem. 2016, 138, 204–221.
- Colucci-D’Amato, L.; Speranza, L.; Volpicelli, F. Neurotrophic Factor BDNF, Physiological Functions and Therapeutic Potential in Depression, Neurodegeneration and Brain Cancer. Int. J. Mol. Sci. 2020, 21, 7777.
- Baydyuk, M.; Xu, B. BDNF signaling and survival of striatal neurons. Front. Cell. Neurosci. 2014, 8, 254.
- Wang, C.S.; Kavalali, E.T.; Monteggia, L.M. BDNF signaling in context: From synaptic regulation to psychiatric disorders. Cell 2022, 185, 62–76.
- Ying, S.W.; Futter, M.; Rosenblum, K.; Webber, M.J.; Hunt, S.P.; Bliss, T.V.; Bramham, C.R. Brain-derived neurotrophic factor induces long-term potentiation in intact adult hippocampus: Requirement for ERK activation coupled to CREB and upregulation of Arc synthesis. J. Neurosci. 2002, 22, 1532–1540.
- Rex, C.S.; Lin, C.Y.; Kramár, E.A.; Chen, L.Y.; Gall, C.M.; Lynch, G. Brain-derived neurotrophic factor promotes long-term potentiation-related cytoskeletal changes in adult hippocampus. J. Neurosci. 2007, 27, 3017–3029.
- Lau, A.G.; Irier, H.A.; Gu, J.; Tian, D.; Ku, L.; Liu, G.; Xia, M.; Fritsch, B.; Zheng, J.Q.; Dingledine, R.; et al. Distinct 3′UTRs differentially regulate activity-dependent translation of brain-derived neurotrophic factor (BDNF). Proc. Natl. Acad. Sci. USA 2010, 107, 15945–15950.
- Pruunsild, P.; Kazantseva, A.; Aid, T.; Palm, K.; Timmusk, T. Dissecting the human BDNF locus: Bidirectional transcription, complex splicing, and multiple promoters. Genomics 2007, 90, 397–406.
- Kowiański, P.; Lietzau, G.; Czuba, E.; Waśkow, M.; Steliga, A.; Moryś, J. BDNF: A Key Factor with Multipotent Impact on Brain Signaling and Synaptic Plasticity. Cell. Mol. Neurobiol. 2018, 38, 579–593.
- Karlović, D.; Serretti, A.; Jevtović, S.; Vrkić, N.; Serić, V.; Peleš, A.M. Diagnostic accuracy of serum brain derived neurotrophic factor concentration in antidepressant naïve patients with first major depression episode. J. Psychiatr. Res. 2013, 47, 162–167.
- Molendijk, M.L.; Bus, B.A.; Spinhoven, P.; Penninx, B.W.; Kenis, G.; Prickaerts, J.; Voshaar, R.C.; Elzinga, B.M. Serum levels of brain-derived neurotrophic factor in major depressive disorder: State-trait issues, clinical features and pharmacological treatment. Mol. Psychiatry 2011, 16, 1088–1095.
- Miyanishi, H.; Nitta, A. A Role of BDNF in the Depression Pathogenesis and a Potential Target as Antidepressant: The Modulator of Stress Sensitivity “Shati/Nat8l-BDNF System” in the Dorsal Striatum. Pharmaceuticals 2021, 14, 889.
- Sakata, K.; Woo, N.H.; Martinowich, K.; Greene, J.S.; Schloesser, R.J.; Shen, L.; Lu, B. Critical role of promoter IV-driven BDNF transcription in GABAergic transmission and synaptic plasticity in the prefrontal cortex. Proc. Natl. Acad. Sci. USA 2009, 106, 5942–5947.
- Sakata, K.; Duke, S.M. Lack of BDNF expression through promoter IV disturbs expression of monoamine genes in the frontal cortex and hippocampus. Neuroscience 2014, 260, 265–275.
- Sakata, K.; Jin, L.; Jha, S. Lack of promoter IV-driven BDNF transcription results in depression-like behavior. Genes Brain Behav. 2010, 9, 712–721.
- Lieb, K.; Dreimüller, N.; Wagner, S.; Schlicht, K.; Falter, T.; Neyazi, A.; Müller-Engling, L.; Bleich, S.; Tadić, A.; Frieling, H. BDNF Plasma Levels and BDNF Exon IV Promoter Methylation as Predictors for Antidepressant Treatment Response. Front. Psychiatry 2018, 9, 511.
- Jevtović, S.; Karlović, D.; Mihaljević-Peleš, A.; Šerić, V.; Vrkić, N.; Jakšić, N. Serum Brain-derived neurotrophic factor (BDNF): The severity and symptomatic dimensions of depression. Psychiatr. Danub. 2011, 23, 363–369.
- Kreinin, A.; Lisson, S.; Nesher, E.; Schneider, J.; Bergman, J.; Farhat, K.; Farah, J.; Lejbkowicz, F.; Yadid, G.; Raskin, L.; et al. Blood BDNF level is gender specific in severe depression. PLoS ONE 2015, 10, e0127643.
- Caldieraro, M.A.; Vares, E.A.; Souza, L.H.; Spanemberg, L.; Guerra, T.A.; Wollenhaupt-Aguiar, B.; Ferrari, P.; Nierenberg, A.A.; Fleck, M.P. Illness severity and biomarkers in depression: Using a unidimensional rating scale to examine BDNF. Compr. Psychiatry 2017, 75, 46–52.
- Katsuki, A.; Yoshimura, R.; Kishi, T.; Hori, H.; Umene-Nakano, W.; Ikenouchi-Sugita, A.; Hayashi, K.; Atake, K.; Iwata, N.; Nakamura, J. Serum levels of brain-derived neurotrophic factor (BDNF), BDNF gene Val66Met polymorphism, or plasma catecholamine metabolites, and response to mirtazapine in Japanese patients with major depressive disorder (MDD). CNS Spectr. 2012, 17, 155–163.
- Brunoni, A.R.; Lopes, M.; Fregni, F. A systematic review and meta-analysis of clinical studies on major depression and BDNF levels: Implications for the role of neuroplasticity in depression. Int. J. Neuropsychopharmacol. 2008, 11, 1169–1180.
- Martinac, M.; Pehar, D.; Karlović, D.; Babić, D.; Marcinko, D.; Jakovljević, M. Metabolic syndrome, activity of the hypothalamic-pituitary-adrenal axis and inflammatory mediators in depressive disorder. Acta Clin. Croat. 2014, 53, 55–71.
- Varghese, F.P.; Brown, E.S. The Hypothalamic-Pituitary-Adrenal Axis in Major Depressive Disorder: A Brief Primer for Primary Care Physicians. Prim. Care Companion J. Clin. Psychiatry 2001, 3, 151–155.
- Iob, E.; Kirschbaum, C.; Steptoe, A. Persistent depressive symptoms, HPA-axis hyperactivity, and inflammation: The role of cognitive-affective and somatic symptoms. Mol. Psychiatry 2020, 25, 1130–1140.
- Zhu, L.J.; Liu, M.Y.; Li, H.; Liu, X.; Chen, C.; Han, Z.; Wu, H.Y.; Jing, X.; Zhou, H.H.; Suh, H.; et al. The different roles of glucocorticoids in the hippocampus and hypothalamus in chronic stress-induced HPA axis hyperactivity. PLoS ONE 2014, 9, e97689.
- Kino, T. Stress, glucocorticoid hormones, and hippocampal neural progenitor cells: Implications to mood disorders. Front. Physiol. 2015, 6, 230.
- Cole, A.B.; Montgomery, K.; Bale, T.L.; Thompson, S.M. What the hippocampus tells the HPA axis: Hippocampal output attenuates acute stress responses via disynaptic inhibition of CRF+ PVN neurons. Neurobiol. Stress 2022, 20, 100473.
- Keller, J.; Gomez, R.; Williams, G.; Lembke, A.; Lazzeroni, L.; Murphy, G.M., Jr.; Schatzberg, A.F. HPA axis in major depression: Cortisol, clinical symptomatology and genetic variation predict cognition. Mol. Psychiatry 2017, 22, 527–536.
- Gjerstad, J.K.; Lightman, S.L.; Spiga, F. Role of glucocorticoid negative feedback in the regulation of HPA axis pulsatility. Stress 2018, 21, 403–416.
- Menke, A. Is the HPA Axis as Target for Depression Outdated, or Is There a New Hope? Front. Psychiatry 2019, 10, 101.
- McEwen, B.S. Glucocorticoids, depression, and mood disorders: Structural remodeling in the brain. Metabolism 2005, 54 (Suppl. S1), 20–23.
- Krugers, H.J.; Lucassen, P.J.; Karst, H.; Joëls, M. Chronic stress effects on hippocampal structure and synaptic function: Relevance for depression and normalization by anti-glucocorticoid treatment. Front. Synaptic Neurosci. 2010, 2, 24.
- Liston, C.; Gan, W.B. Glucocorticoids are critical regulators of dendritic spine development and plasticity in vivo. Proc. Natl. Acad. Sci. USA 2011, 108, 16074–16079.
- Tata, D.A.; Anderson, B.J. The effects of chronic glucocorticoid exposure on dendritic length, synapse numbers and glial volume in animal models: Implications for hippocampal volume reductions in depression. Physiol. Behav. 2010, 99, 186–193.
- Bhatt, S.; Nagappa, A.N.; Patil, C.R. Role of oxidative stress in depression. Drug Discover. Today 2020, 25, 1270–1276.
- Juszczyk, G.; Mikulska, J.; Kasperek, K.; Pietrzak, D.; Mrozek, W.; Herbet, M. Chronic Stress and Oxidative Stress as Common Factors of the Pathogenesis of Depression and Alzheimer’s Disease: The Role of Antioxidants in Prevention and Treatment. Antioxidants 2021, 10, 1439.
- Salim, S. Oxidative Stress and the Central Nervous System. J. Pharmacol. Exp. Ther. 2017, 360, 201–205.
- Wang, X.; Michaelis, E.K. Selective neuronal vulnerability to oxidative stress in the brain. Front. Aging Neurosci. 2010, 2, 12.
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules 2019, 24, 1583.
- Lee, K.H.; Cha, M.; Lee, B.H. Neuroprotective Effect of Antioxidants in the Brain. Int. J. Mol. Sci. 2020, 21, 7152.
- Cobley, J.N.; Fiorello, M.L.; Bailey, D.M. 13 reasons why the brain is susceptible to oxidative stress. Redox Biol. 2018, 15, 490–503.
- Bakunina, N.; Pariante, C.M.; Zunszain, P.A. Immune mechanisms linked to depression via oxidative stress and neuroprogression. Immunology 2015, 144, 365–373.
- Morris, G.; Berk, M.; Klein, H.; Walder, K.; Galecki, P.; Maes, M. Nitrosative Stress, Hypernitrosylation, and Autoimmune Responses to Nitrosylated Proteins: New Pathways in Neuroprogressive Disorders Including Depression and Chronic Fatigue Syndrome. Mol. Neurobiol. 2017, 54, 4271–4291.
- Lopresti, A.L.; Maker, G.L.; Hood, S.D.; Drummond, P.D. A review of peripheral biomarkers in major depression: The potential of inflammatory and oxidative stress biomarkers. Prog. Neuropsychopharmacol. Biol. Psychiatry 2014, 48, 102–111.
- Correia, A.S.; Cardoso, A.; Vale, N. Oxidative Stress in Depression: The Link with the Stress Response, Neuroinflammation, Serotonin, Neurogenesis and Synaptic Plasticity. Antioxidants 2023, 12, 470.
- Zhong, J.; Li, G.; Xu, H.; Wang, Y.; Shi, M. Baicalin ameliorates chronic mild stress-induced depression-like behaviors in mice and attenuates inflammatory cytokines and oxidative stress. Braz. J. Med. Biol. Res. 2019, 52, e8434.
- Vaváková, M.; Ďuračková, Z.; Trebatická, J. Markers of Oxidative Stress and Neuroprogression in Depression Disorder. Oxid. Med. Cell. Longev. 2015, 2015, 898393.
- Forlenza, M.J.; Miller, G.E. Increased serum levels of 8-hydroxy-2′-deoxyguanosine in clinical depression. Psychosom. Med. 2006, 68, 1–7.
- Lindqvist, D.; Dhabhar, F.S.; James, S.J.; Hough, C.M.; Jain, F.A.; Bersani, F.S.; Reus, V.I.; Verhoeven, J.E.; Epel, E.S.; Mahan, L.; et al. Oxidative stress, inflammation and treatment response in major depression. Psychoneuroendocrinology 2017, 76, 197–205.
- Mazereeuw, G.; Herrmann, N.; Andreazza, A.C.; Khan, M.M.; Lanctôt, K.L. A meta-analysis of lipid peroxidation markers in major depression. Neuropsychiatr. Dis. Treat. 2015, 11, 2479–2491.
- Liu, T.; Zhong, S.; Liao, X.; Chen, J.; He, T.; Lai, S. A Meta-Analysis of Oxidative Stress Markers in Depression. PLoS ONE 2015, 10, e0138904.
- Islam, M.R.; Islam, M.R.; Ahmed, I.; Moktadir, A.A.; Nahar, Z.; Islam, M.S.; Shahid, S.F.B.; Islam, S.N.; Islam, M.S.; Hasnat, A. Elevated serum levels of malondialdehyde and cortisol are associated with major depressive disorder: A case-control study. SAGE Open Med. 2018, 6, 2050312118773953.
- Nobis, A.; Zalewski, D.; Waszkiewicz, N. Peripheral Markers of Depression. J. Clin. Med. 2020, 9, 3793.
- Camkurt, M.A.; Fındıklı, E.; İzci, F.; Kurutaş, E.B.; Tuman, T.C. Evaluation of malondialdehyde, superoxide dismutase and catalase activity and their diagnostic value in drug naïve, first episode, non-smoker major depression patients and healthy controls. Psychiatry Res. 2016, 238, 81–85.
- Michel, T.M.; Camara, S.; Tatschner, T.; Frangou, S.; Sheldrick, A.J.; Riederer, P.; Grünblatt, E. Increased xanthine oxidase in the thalamus and putamen in depression. World J. Biol. Psychiatry 2010, 11 Pt 2, 314–320.
- Xu, M.; Tian, P.; Zhu, H.; Zou, R.; Zhao, J.; Zhang, H.; Wang, G.; Chen, W. Lactobacillus paracasei CCFM1229 and Lactobacillus rhamnosus CCFM1228 Alleviated Depression- and Anxiety-Related Symptoms of Chronic Stress-Induced Depression in Mice by Regulating Xanthine Oxidase Activity in the Brain. Nutrients 2022, 14, 1294.
- Sarandol, A.; Sarandol, E.; Eker, S.S.; Erdinc, S.; Vatansever, E.; Kirli, S. Major depressive disorder is accompanied with oxidative stress: Short-term antidepressant treatment does not alter oxidative-antioxidative systems. Hum. Psychopharmacol. 2007, 22, 67–73.
- Katrenčíková, B.; Vaváková, M.; Paduchová, Z.; Nagyová, Z.; Garaiova, I.; Muchová, J.; Ďuračková, Z.; Trebatická, J. Oxidative Stress Markers and Antioxidant Enzymes in Children and Adolescents with Depressive Disorder and Impact of Omega-3 Fatty Acids in Randomised Clinical Trial. Antioxidants 2021, 10, 1256.
- Tsai, M.C.; Huang, T.L. Increased activities of both superoxide dismutase and catalase were indicators of acute depressive episodes in patients with major depressive disorder. Psychiatry Res. 2016, 235, 38–42.
- Sabade, S.B.; Gagare, D.B.; Vikhe, B.B.; Bhalerao, M.M. Study of serum catalase in depression at Pravara institute of medical sciences. Indian J. Clin. Anat. Physiol. 2022, 9, 279–282.
- Maes, M.; Mihaylova, I.; Kubera, M.; Uytterhoeven, M.; Vrydags, N.; Bosmans, E. Lower whole blood glutathione peroxidase (GPX) activity in depression, but not in myalgic encephalomyelitis/chronic fatigue syndrome: Another pathway that may be associated with coronary artery disease and neuroprogression in depression. Neuro. Endocrinol. Lett. 2011, 32, 133–140.
- Bilici, M.; Efe, H.; Köroğlu, M.A.; Uydu, H.A.; Bekaroğlu, M.; Değer, O. Antioxidative enzyme activities and lipid peroxidation in major depression: Alterations by antidepressant treatments. J. Affect. Disord. 2001, 64, 43–51.
- da Cruz Jung, I.E.; da Cruz, I.B.M.; Barbisan, F.; Trott, A.; Houenou, L.J.; Osmarin Turra, B.; Duarte, T.; de Souza Praia, R.; Maia-Ribeiro, E.A.; da Costa Escobar Piccoli, J.; et al. Superoxide imbalance triggered by Val16Ala-SOD2 polymorphism increases the risk of depression and self-reported psychological stress in free-living elderly people. Mol. Gen. Genom. Med. 2019, 8, e1080.
- Wigner, P.; Czarny, P.; Synowiec, E.; Bijak, M.; Białek, K.; Talarowska, M.; Galecki, P.; Szemraj, J.; Sliwinski, T. Variation of genes involved in oxidative and nitrosative stresses in depression. Eur. Psychiatry 2018, 48, 38–48.
- Huang, Q.; Liu, H.; Suzuki, K.; Ma, S.; Liu, C. Linking What We Eat to Our Mood: A Review of Diet, Dietary Antioxidants, and Depression. Antioxidants 2019, 8, 376.
- Maes, M.; De Vos, N.; Pioli, R.; Demedts, P.; Wauters, A.; Neels, H.; Christophe, A. Lower serum vitamin E concentrations in major depression. Another marker of lowered antioxidant defenses in that illness. J. Affect. Disord. 2000, 58, 241–246.
- Gautam, M.; Agrawal, M.; Gautam, M.; Sharma, P.; Gautam, A.S.; Gautam, S. Role of antioxidants in generalised anxiety disorder and depression. Indian J. Psychiatry 2012, 54, 244–247.
- Moylan, S.; Berk, M.; Dean, O.M.; Samuni, Y.; Williams, L.J.; O’Neil, A.; Hayley, A.C.; Pasco, J.A.; Anderson, G.; Jacka, F.N.; et al. Oxidative & nitrosative stress in depression: Why so much stress? Neurosci. Biobehav. Rev. 2014, 45, 46–62.
- Abshirini, M.; Siassi, F.; Koohdani, F.; Qorbani, M.; Mozaffari, H.; Aslani, Z.; Soleymani, M.; Entezarian, M.; Sotoudeh, G. Dietary total antioxidant capacity is inversely associated with depression, anxiety and some oxidative stress biomarkers in postmenopausal women: A cross-sectional study. Ann. Gen. Psychiatry 2019, 18, 3.
- Wu, S.-X.; Li, J.; Zhou, D.-D.; Xiong, R.-G.; Huang, S.-Y.; Saimaiti, A.; Shang, A.; Li, H.-B. Possible Effects and Mechanisms of Dietary Natural Products and Nutrients on Depression and Anxiety: A Narrative Review. Antioxidants 2022, 11, 2132.
- Winiarska-Mieczan, A.; Kwiecień, M.; Jachimowicz-Rogowska, K.; Donaldson, J.; Tomaszewska, E.; Baranowska-Wójcik, E. Anti-Inflammatory, Antioxidant, and Neuroprotective Effects of Polyphenols—Polyphenols as an Element of Diet Therapy in Depressive Disorders. Int. J. Mol. Sci. 2023, 24, 2258.
- Moritz, B.; Schmitz, A.E.; Rodrigues, A.L.S.; Dafre, A.L.; Cunha, M.P. The role of vitamin C in stress-related disorders. J. Nutr. Biochem. 2020, 85, 108459.
- Yosaee, S.; Keshtkaran, Z.; Abdollahi, S.; Shidfar, F.; Sarris, J.; Soltani, S. The effect of vitamin C supplementation on mood status in adults: A systematic review and meta-analysis of randomized controlled clinical trials. Gen. Hosp. Psychiatry 2021, 71, 36–42.
- Sahraian, A.; Ghanizadeh, A.; Kazemeini, F. Vitamin C as an adjuvant for treating major depressive disorder and suicidal behavior, a randomized placebo-controlled clinical trial. Trials 2015, 16, 94.
- Amr, M.; El-Mogy, A.; Shams, T.; Vieira, K.; Lakhan, S.E. Efficacy of vitamin C as an adjunct to fluoxetine therapy in pediatric major depressive disorder: A randomized, double-blind, placebo-controlled pilot study. Nutr. J. 2013, 12, 31.
- Manosso, L.M.; Camargo, A.; Dafre, A.L.; Rodrigues, A.L.S. Vitamin E for the management of major depressive disorder: Possible role of the anti-inflammatory and antioxidant systems. Nutr. Neurosci. 2020, 25, 1310–1324.
- Lee, A.R.Y.B.; Tariq, A.; Lau, G.; Tok, N.W.K.; Tam, W.W.S.; Ho, C.S.H. Vitamin E, Alpha-Tocopherol, and Its Effects on Depression and Anxiety: A Systematic Review and Meta-Analysis. Nutrients 2022, 14, 656.
- Berk, M.; Dean, O.M.; Cotton, S.M.; Jeavons, S.; Tanious, M.; Kohlmann, K.; Hewitt, K.; Moss, K.; Allwang, C.; Schapkaitz, I.; et al. The efficacy of adjunctive N-acetylcysteine in major depressive disorder: A double-blind, randomized, placebo-controlled trial. J. Clin. Psychiatry 2014, 75, 628–636.
- Fernandes, B.S.; Dean, O.M.; Dodd, S.; Malhi, G.S.; Berk, M. N-Acetylcysteine in depressive symptoms and functionality: A systematic review and meta-analysis. J. Clin. Psychiatry 2016, 77, e457–e466.
- Ooi, S.L.; Green, R.; Pak, S.C. N-Acetylcysteine for the Treatment of Psychiatric Disorders: A Review of Current Evidence. Biomed. Res. Int. 2018, 2469486.
- Woo, J.; Cho, H.; Seol, Y.; Kim, S.H.; Park, C.; Yousefian-Jazi, A.; Hyeon, S.J.; Lee, J.; Ryu, H. Power Failure of Mitochondria and Oxidative Stress in Neurodegeneration and Its Computational Models. Antioxidants 2021, 10, 229.
- Tsang, Y.-L.; Kao, C.-L.; Lin, S.-C.A.; Li, C.-J. Mitochondrial Dysfunction and Oxidative Stress in Aging and Disease. Biomedicines 2022, 10, 2872.
- Wang, Q.; Dwivedi, Y. Transcriptional profiling of mitochondria associated genes in prefrontal cortex of subjects with major depressive disorder. World J. Biol. Psychiatry 2017, 18, 592–603.
- Allen, J.; Romay-Tallon, R.; Brymer, K.J.; Caruncho, H.J.; Kalynchuk, L.E. Mitochondria and Mood: Mitochondrial Dysfunction as a Key Player in the Manifestation of Depression. Front. Neurosci. 2018, 12, 386.
- Tobe, E.H. Mitochondrial dysfunction, oxidative stress, and major depressive disorder. Neuropsychiatr. Dis. Treat. 2013, 9, 567–573.
- Bansal, Y.; Kuhad, A. Mitochondrial Dysfunction in Depression. Curr. Neuropharmacol. 2016, 14, 610–618.
- Lindqvist, D.; Wolkowitz, O.M.; Picard, M.; Ohlsson, L.; Bersani, F.S.; Fernström, J.; Westrin, Å.; Hough, C.M.; Lin, J.; Reus, V.I.; et al. Circulating cell-free mitochondrial DNA, but not leukocyte mitochondrial DNA copy number, is elevated in major depressive disorder. Neuropsychopharmacology 2018, 43, 1557–1564.
- Meyer, J.H.; Ginovart, N.; Boovariwala, A.; Sagrati, S.; Hussey, D.; Garcia, A.; Young, T.; Praschak-Rieder, N.; Wilson, A.A.; Houle, S. Elevated monoamine oxidase a levels in the brain: An explanation for the monoamine imbalance of major depression. Arch. Gen. Psychiatry 2006, 63, 1209–1216.
- Chiuccariello, L.; Houle, S.; Miler, L.; Cooke, R.G.; Rusjan, P.M.; Rajkowska, G.; Levitan, R.D.; Kish, S.J.; Kolla, N.J.; Ou, X.; et al. Elevated monoamine oxidase a binding during major depressive episodes is associated with greater severity and reversed neurovegetative symptoms. Neuropsychopharmacology 2014, 39, 973–980.
- Naoi, M.; Maruyama, W.; Shamoto-Nagai, M. Type A monoamine oxidase and serotonin are coordinately involved in depressive disorders: From neurotransmitter imbalance to impaired neurogenesis. J. Neural. Transm. 2018, 125, 53–66.
- Stahl, S.M.; Felker, A. Monoamine oxidase inhibitors: A modern guide to an unrequited class of antidepressants. CNS Spectr. 2008, 13, 855–870.
- Flockhart, D.A. Dietary restrictions and drug interactions with monoamine oxidase inhibitors: An update. J. Clin. Psychiatry 2012, 73 (Suppl. S1), 17–24.
- Shulman, K.I.; Herrmann, N.; Walker, S.E. Current place of monoamine oxidase inhibitors in the treatment of depression. CNS Drugs 2013, 27, 789–797.
- Haroon, E.; Miller, A.H.; Sanacora, G. Inflammation, Glutamate, and Glia: A Trio of Trouble in Mood Disorders. Neuropsychopharmacology 2017, 42, 193–215.
- Jazvinšćak Jembrek, M.; Radovanović, V.; Vlainić, J.; Vuković, L.; Hanžić, N. Neuroprotective effect of zolpidem against glutamate-induced toxicity is mediated via the PI3K/Akt pathway and inhibited by PK11195. Toxicology 2018, 406–407, 58–69.
- Olloquequi, J.; Cornejo-Córdova, E.; Verdaguer, E.; Soriano, F.X.; Binvignat, O.; Auladell, C.; Camins, A. Excitotoxicity in the pathogenesis of neurological and psychiatric disorders: Therapeutic implications. J. Psychopharmacol. 2018, 32, 265–275.
- Kuhn, D.M.; Geddes, T.J. Peroxynitrite inactivates tryptophan hydroxylase via sulfhydryl oxidation. Coincident nitration of enzyme tyrosyl residues has minimal impact on catalytic activity. J. Biol. Chem. 1999, 274, 29726–29732.
- Wang, H.; He, Y.; Sun, Z.; Ren, S.; Liu, M.; Wang, G.; Yang, J. Microglia in depression: An overview of microglia in the pathogenesis and treatment of depression. J. Neuroinflamm. 2022, 19, 132.
- Atagun, M.İ.; Atay, O.C.; Balaban, O.D.; Ipekcioglu, D.; Alpugan, B.; Yalcin, S.; Senat, A.; Karamustafalioglu, N.; Ilnem, M.C.; Erel, O. Serum nitric oxide levels are depleted in depressed patients treated with electroconvulsive therapy. Indian J. Psychiatry 2021, 63, 456–461.
- Savitz, J. Role of Kynurenine Metabolism Pathway Activation in Major Depressive Disorders. Curr. Top. Behav. Neurrosci. 2017, 31, 249–267.
- Marx, W.; McGuinness, A.J.; Rocks, T.; Ruusunen, A.; Cleminson, J.; Walker, A.J.; Gomes-da-Costa, S.; Lane, M.; Sanches, M.; Diaz, A.P.; et al. The kynurenine pathway in major depressive disorder, bipolar disorder, and schizophrenia: A meta-analysis of 101 studies. Mol. Psychiatry 2021, 26, 4158–4178.
- Hacioglu, G.; Senturk, A.; Ince, I.; Alver, A. Assessment of oxidative stress parameters of brain-derived neurotrophic factor heterozygous mice in acute stress model. Iran. J. Basic Med. Sci. 2016, 19, 388–393.
- Afridi, R.; Suk, K. Neuroinflammatory Basis of Depression: Learning from Experimental Models. Front. Cell. Neurosci. 2021, 15, 691067.
- Crnković, D.; Buljan, D.; Karlović, D.; Krmek, M. Connection between inflammatory markers, antidepressants and depression. Acta Clin. Croat. 2012, 51, 25–33.
- Berk, M.; Williams, L.J.; Jacka, F.N.; O’Neil, A.; Pasco, J.A.; Moylan, S.; Allen, N.B.; Stuart, A.L.; Hayley, A.C.; Byrne, M.L.; et al. So depression is an inflammatory disease, but where does the inflammation come from? BMC Med. 2013, 11, 200.
- Himmerich, H.; Patsalos, O.; Lichtblau, N.; Ibrahim, M.A.A.; Dalton, B. Cytokine Research in Depression: Principles, Challenges, and Open Questions. Front. Psychiatry 2019, 10, 30.
- Beurel, E.; Toups, M.; Nemeroff, C.B. The Bidirectional Relationship of Depression and Inflammation: Double Trouble. Neuron 2020, 107, 234–256.
- Kim, I.-B.; Lee, J.-H.; Park, S.-C. The Relationship between Stress, Inflammation, and Depression. Biomedicines 2022, 10, 1929.
- Lee, C.H.; Giuliani, F. The Role of Inflammation in Depression and Fatigue. Front. Immunol. 2019, 10, 1696.
- Hannestad, J.; DellaGioia, N.; Bloch, M. The effect of antidepressant medication treatment on serum levels of inflammatory cytokines: A meta-analysis. Neuropsychopharmacology 2011, 36, 2452–2459.
- Felger, J.C.; Lotrich, F.E. Inflammatory cytokines in depression: Neurobiological mechanisms and therapeutic implications. Neuroscience 2013, 246, 199–229.
- Capuron, L.; Miller, A.H. Immune system to brain signaling: Neuropsychopharmacological implications. Pharmacol. Ther. 2011, 130, 226–238.
- Jeenger, J.; Singroha, V.; Sharma, M.; Mathur, D.; Mathur, D.M. C-reactive protein, brain-derived neurotrophic factor, interleukin-2, and stressful life events in drug-naive first-episode and recurrent depression: A cross-sectional study. Indian J. Psychiatry 2018, 60, 334–339.
- Rengasamy, M.; Marsland, A.; Spada, M.; Hsiung, K.; Kovats, T.; Price, R.B. A chicken and egg scenario in psychoneuroimmunology: Bidirectional linking cytokines and depression. J. Affect. Disord. Rep. 2021, 6, 100177.
- Zunszain, P.A.; Anacker, C.; Cattaneo, A.; Carvalho, L.A.; Pariante, C.M. Glucocorticoids, cytokines and brain abnormalities in depression. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2011, 35, 722–729.
- Karlović, D.; Serretti, A.; Vrkić, N.; Martinac, M.; Marčinko, D. Serum concentrations of CRP, IL-6, TNF-α and cortisol in major depressive disorder with melancholic or atypical features. Psychiatry Res. 2012, 198, 74–80.
- Leonard, B.E. The immune system, depression and the action of antidepressants. Prog. Neuropsychopharmacol. Biol. Psychiatry 2001, 25, 767–780.
- Zhang, J.; Wang, X.; Vikash, V.; Ye, Q.; Wu, D.; Liu, Y.; Dong, W. ROS and ROS-Mediated Cellular Signaling. Oxid. Med. Cell. Longev. 2016, 2016, 4350965.
- Simpson, D.S.A.; Oliver, P.L. ROS Generation in Microglia: Understanding Oxidative Stress and Inflammation in Neurodegenerative Disease. Antioxidants 2020, 9, 743.
- Lee, K.H.; Cha, M.; Lee, B.H. Crosstalk between Neuron and Glial Cells in Oxidative Injury and Neuroprotection. Int. J. Mol. Sci. 2021, 22, 13315.
- Koo, J.W.; Russo, S.J.; Ferguson, D.; Nestler, E.J.; Duman, R.S. Nuclear factor-kappaB is a critical mediator of stress-impaired neurogenesis and depressive behavior. Proc. Natl. Acad. Sci. USA 2010, 107, 2669–2674.
- Li, Y.; Song, W.; Tong, Y.; Zhang, X.; Zhao, J.; Gao, X.; Yong, J.; Wang, H. Isoliquiritin ameliorates depression by suppressing NLRP3-mediated pyroptosis via miRNA-27a/SYK/NF-κB axis. J. Neuroinflamm. 2021, 18, 1.
- Wang, Y.; Xu, J.; Liu, Y.; Li, Z.; Li, X. TLR4-NF-κB Signal Involved in Depressive-like Behaviors and Cytokine Expression of Frontal Cortex and Hippocampus in Stressed C57BL/6 and ob/ob Mice. Neural Plast. 2018, 2018, 7254016.
- Chen, H.; Ma, Y.; Chen, M.; Chen, J.; Chen, J. Safflower extract improves depression in mice by inhibiting the TLR4-NLRP3 inflammation signaling pathway. Ann. Palliat. Med. 2021, 10, 8015–8023.
- Nazari, M.; Khodadadi, H.; Fathalizadeh, J.; Hassanshahi, G.; Bidaki, R.; Ayoobi, F.; Hajebrahimi, B.; Bagheri, F.; Arababadi, M.K. Defective NF-kB transcription factor as the mediator of inflammatory responses: A study on depressed Iranian medical students. Clin. Lab. 2013, 59, 827–830.
- Miklowitz, D.J.; Portnoff, L.C.; Armstrong, C.C.; Keenan-Miller, D.; Breen, E.C.; Muscatell, K.A.; Eisenberger, N.I.; Irwin, M.R. Inflammatory cytokines and nuclear factor-kappa B activation in adolescents with bipolar and major depressive disorders. Psychiatry Res. 2016, 241, 315–322.
- Müller, N. COX-2 Inhibitors, Aspirin, and Other Potential Anti-Inflammatory Treatments for Psychiatric Disorders. Front. Psychiatry 2019, 10, 375.
- Song, Q.; Feng, Y.B.; Wang, L.; Shen, J.; Li, Y.; Fan, C.; Wang, P.; Yu, S.Y. COX-2 inhibition rescues depression-like behaviors via suppressing glial activation, oxidative stress and neuronal apoptosis in rats. Neuropharmacology 2019, 160, 107779.
- He, Y.; Han, Y.; Liao, X.; Zou, M.; Wang, Y. Biology of cyclooxygenase-2: An application in depression therapeutics. Front. Psychiatry 2022, 13, 1037588.
- Gałecki, P.; Florkowski, A.; Bieńkiewicz, M.; Szemraj, J. Functional polymorphism of cyclooxygenase-2 gene (G-765C) in depressive patients. Neuropsychobiology 2010, 62, 116–120.
- Seo, J.S.; Park, J.Y.; Choi, J.; Kim, T.K.; Shin, J.H.; Lee, J.K.; Han, P.L. NADPH oxidase mediates depressive behavior induced by chronic stress in mice. J. Neurosci. 2012, 32, 9690–9699.
More