Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Ramy Rahmé and Version 2 by Rita Xu.
TP53 mutations are less frequent in myelodysplastic syndromes (MDS) and acute myeloid leukemia (AML) than in solid tumors, except in secondary and therapy-related MDS/AMLs, and in cases with complex monosomal karyotype. As in solid tumors, missense mutations predominate, with the same hotspot mutated codons (particularly codons 175, 248, 273). As TP53-mutated MDS/AMLs are generally associated with complex chromosomal abnormalities, it is not always clear when TP53 mutations occur in the pathophysiological process.
- mutant p53
- cancer
- myelodysplasia
- acute myeloid leukemia
1. Introduction
The transcription factor p53, encoded by the TP53 gene in humans, is one of the most studied tumor suppressor proteins. In normal cells, p53 activity is low, but in response to DNA damage and numerous other stress signals, p53 levels rise dramatically and result in the activation and transcription of genes with important roles in cell cycle arrest, senescence, apoptosis, metabolism, and differentiation [1]. The sum of these activities is to ensure that an abnormal cell fails to arise and proliferate. During normal hematopoiesis, p53 activities preserve genome integrity and regulate several cellular processes that maintain the normal stem cell pool and serve as a barrier to tumorigenesis [2][3][2,3].
Perturbations in p53 activity or p53-dependent pathways are required for the development of most cancers [4], and there is evidence in many situations to suggest that the restoration or reactivation of physiological p53 function may be therapeutic [5][6][7][8][5,6,7,8]. Various mechanisms are responsible for the disruption of p53 activity in cancer, mainly deletion or mutation of the TP53 gene, and overexpression of the p53 negative regulators Mdm2 and Mdm4 [9][10][11][9,10,11]. Several isoforms are encoded by the TP53 gene and appear to play different roles in tumorigenesis/cancer progression [12] or response to treatment, for example in acute myeloid leukemia (AML) [13]. Regardless of the mechanism behind p53 dysfunction, the downstream consequences are profound due to the very large spectrum of biological activities in which p53 is normally implicated. The clinical correlation between p53 mutational status in cancer cells and resistance to treatment has been studied since the 1990s. In breast cancer, presence of a TP53 mutation is associated with resistance to doxorubicin [14][15][14,15]. Similarly, ovarian cancer patients harboring a TP53 mutation are less sensitive to treatment with cisplatin [16][17][16,17]. Moreover, the association of TP53 mutation with chemoresistance and poor prognosis has also been observed in lung [18], gastric, and colorectal cancers [19], as well as in hematological malignancies [20][21][20,21]. Apart from triggering chemoresistance, p53 mutants are also able to attenuate cancer response to radiotherapy [14][19][22][14,19,22].
Myelodysplastic syndromes (MDS) are a clonal hematopoietic stem cell disorders characterized by cell dysplasia, ineffective hematopoiesis that leads to cytopenias (mainly anemia) and a variable risk of progression to AML [23]. Furthermore, AML is characterized by clonal expansion of undifferentiated myeloid precursors, resulting in impaired hematopoiesis and life-threatening cytopenias. Among the prognostic factors identified in both malignancies, presence of a mutation in the TP53 gene indicates a particularly dismal prognosis irrespective of the treatment administered [24].
2. Characteristics of TP53 Mutations in MDS/AML
3. When Do TP53 Mutations Arise in MDS and AML Cells?
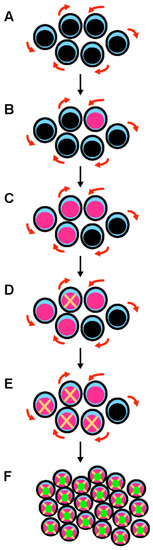
Figure 1. A multistep process leads to the development of TP53-mutated myelodysplastic syndrome and acute myeloid leukemia. (A) Hematopoietic stem cells (HSCs) are the only cells within the hematopoietic system that possess the potential for both multipotency (i.e., the potential to differentiate into all of the mature blood cell type) and self-renewal (i.e., the potential to make more stem cells, thus perpetuating the stem cell pool throughout life). Self-renewal is illustrated with red arrows. (B) A mutation in the TP53 gene can emerge in a hematopoietic stem cell (pink nucleus). This mutation can appear spontaneously and is selected in response to various stresses, including exposure to cytotoxic treatments. This mutation can be induced by these cytotoxic treatments (such as chemotherapy or radiation therapy) or by workplace exposure to toxic chemicals and carcinogenic substances such as benzene. (C) Mutant p53 confers a competitive advantage in the stem cell compartment. At this early stage, cells express both wild type and mutant p53 proteins (potential dominant-negative effect). (D) TP53-mutated HSCs acquire chromosomal changes and gene mutations that either result from mutant p53-related genomic instability and chromothripsis, or are induced by cytotoxic chemotherapy/radiation. (E) These acquired genetic changes further enhance the fitness of TP53-mutated HSCs. (F) Further chromosomal changes lead to the loss of the wild type TP53 allele by 17p deletion or monosomy 17. Loss of wild type TP53 functions and potential gain-of-function activities of mutant p53 are responsible for the development of overt acute myeloid leukemia.
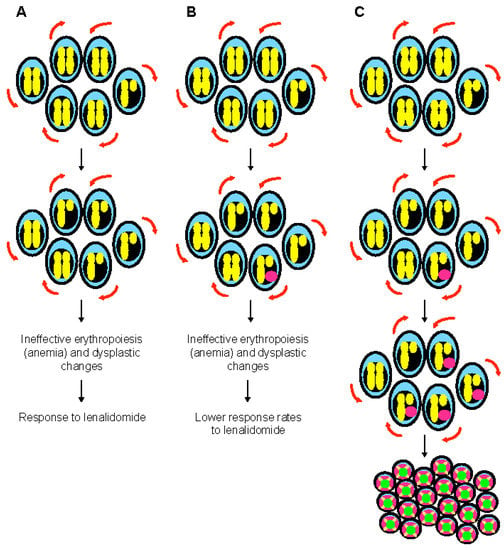
Figure 2. The special case of myelodysplastic syndrome with 5q deletion. (A) Deletion of 5q chromosome is somatically acquired and heterozygous. This chromosomal abnormality is present in the hematopoietic stem cell compartment and can be found in all lineages. The clinical phenotype of 5q- syndrome (i.e., ineffective erythropoiesis and dysplastic changes) is related to haploinsufficient gene expression of several genes such as RPS14, APC, and EGR1. Self-renewal is illustrated with red arrows. (B) Monoallelic TP53 mutations are seen at diagnosis in almost 20% of patients with 5q- syndrome, generally at low variant allelic frequency. These mutations are associated with resistance to lenalidomide, but have generally a limited impact on survival. At this stage, karyotype is non-complex. (C) Monoallelic TP53 mutations are associated with genomic instability and chromothripsis in myelodysplastic syndrome with isolated 5q deletion. Progression to higher risk myelodysplastic syndrome and acute myeloid leukemia is preceded by acquisition of a complex karyotype, including 17p deletion and biallelic TP53 inactivation.