Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Erin Norris and Version 2 by Camila Xu.
The fibrinolytic system plays a crucial role in maintaining vascular integrity and regulating blood clot formation by lysing fibrin clots in both healthy and disease conditions. In addition to hemostasis, the fibrinolytic system also plays crucial roles in wound healing, tissue remodeling, and inflammation. This system is composed of various proteins, including plasminogen, tissue plasminogen activator (tPA), and urokinase plasminogen activator (uPA).
- Alzheimer’s disease
- contact system
- vasculature
1. Plasminogen
Plasmin is a serine protease that is generated from plasminogen by the proteolytic activation of tPA or uPA. Plasminogen is expressed and secreted primarily by hepatocytes in the liver [1][64]. The primary function of plasmin is to lyse fibrin clots, which are formed in response to injury or inflammatory activity. This process is known as fibrinolysis, which is critical for the maintenance of normal blood flow and tissue repair. In addition to its role in blood clot resolution, plasmin is also involved in brain function. In the brain, plasminogen is expressed and secreted by neurons [2][65], and it has been associated with the regulation of synaptic activity by affecting long-term potentiation through the maturation of brain-derived neurotrophic factor (BDNF) [3][66]. Plasminogen’s conversion to plasmin has been implicated in a variety of pathological processes in the brain, including neurodegeneration and neuroinflammation [4][5][67,68].
In AD, it has been suggested that plasmin aids in the clearance of Aβ, as demonstrated by delayed Aβ clearance in plasminogen-deficient mice [6][62]. In the brains of AD mouse models, plasmin and tPA activity are reduced, which could explain the progressive accumulation of parenchymal brain Aβ [6][62]. This finding is partially consistent with human data that show AD brains have low plasmin activity compared to brains from non-demented individuals [7][69]. However, other human studies concluded that plasmin levels/activity are unchanged in AD brains or cerebrospinal fluid (CSF) samples [8][9][70,71]. The inconsistency in plasmin activity in humans may be due to genetic factors, as a reduction in brain plasmin activity was found to correlate with the presence of the APOE epsilon 4 variant in AD patients [10][72]. However, additional studies are needed to confirm this hypothesis. On the other hand, silencing only blood-derived plasminogen using targeted antisense oligonucleotide technology reduces neuroinflammation and Aβ deposition in a mouse model of AD [11][73]. These data suggest that plasminogen may have different effects throughout the body: brain plasminogen appears to be beneficial as it may aid in Aβ clearance, whereas blood-derived plasminogen may be harmful as it can promote proinflammatory cascades that ultimately impair brain function. These findings highlight the importance of considering the location-specific effects of plasminogen in developing therapeutic strategies for AD.
2. Tissue Plasminogen Activator (tPA)
In the vasculature, tPA is primarily expressed and secreted by endothelial cells located along the blood vessel walls. Here, tPA’s critical function is to convert plasminogen to plasmin, aiding in the regulation of blood clot formation and dissolution. In addition to its role in the blood, tPA has important functions in the brain, where it is synthesized and secreted by neurons and some glial cells. In the brain, tPA is involved in a variety of processes, including synaptic plasticity, neuronal survival, and neuroinflammation [12][74]. Overall, tPA is a critical protein involved in the regulation of both the vasculature and the central nervous system, and changes in its levels and activity are associated with different neurological disorders, including AD [4][12][67,74].
With regards to AD, it has been hypothesized by multiple labs that tPA directly participates in Aβ clearance. Studies have shown that tPA interacts with Aβ [13][75] and co-deposits alongside plasminogen with Aβ plaques in the Tg2576 AD mouse model [14][76]. In the hippocampus and amygdala of two AD mouse lines (TgCRND8 and Tg2576), tPA levels and activity are decreased, and injection of Aβ into the hippocampus of tPA-deficient AD mice leads to delayed Aβ clearance. These findings indicate that tPA may be involved in Aβ clearance in the brain, likely through the activation of plasmin [6][13][62,75]. Other groups have corroborated these findings in various other AD mouse lines [15][16][17][18][19][77,78,79,80,81]. For instance, treatment with recombinant tPA reduces Aβ burden in AD mice [17][79], and partial (heterozygous) or complete deficiency (homozygous) of tPA significantly worsens Aβ deposition and overall disease pathology in AD mice [15][77].
tPA may also be involved in CAA pathology in a mouse model of AD. However, the mechanism of how reduced tPA activity in the vasculature promotes CAA may be independent of plasmin and rather via NMDA (N-methyl-D-aspartate) receptor activation and nitric oxide release [18][80]. Interestingly, one study found that enhancing tPA activity by blocking PAI-1 (the main tPA inhibitor in the blood) did not reduce Aβ deposition in the brain but only affected CAA Aβ deposition [18][80]. This result suggests that increasing blood tPA activity reduced levels of Aβ40 but not Aβ42. In another study where brain slices were treated ex vivo, treatment with tPA in combination with plasminogen did not reduce the number of “mature” Aβ plaques, though it did reduce the amount and complexity of Aβ fibrils [14][76]. These data suggest that Aβ structure, fibril length, and aggregate complexity are important factors in the tPA-mediated clearance of Aβ in both blood and the brain. However, further research is needed to better understand tPA’s mechanistic role in the clearance of Aβ, especially with regard to different Aβ mutants and structural species.
Human data supporting the role of tPA in AD is complex and varied. Some studies report decreased tPA activity in AD patient brains or a negative correlation between tPA levels and Aβ load [19][20][81,82], which supports the role of tPA in Aβ clearance. However, another group reports an increase in total tPA protein in AD patients [21][83]. No changes in tPA levels in CSF or plasma have been reported in AD patients compared to non-demented control individuals [9][22][71,84]. However, the tPA/PAI-1 ratio is elevated in serum samples from MCI and AD patients, suggesting a reduction in tPA activity in their blood [23][85].
In addition to Aβ clearance, tPA has been linked to tau pathology in AD. A study using primary hippocampal neurons in culture suggests that tPA can cause tau phosphorylation through an ERK (extracellular signal-regulated kinase) signaling mechanism independent of plasmin [24][63]. Another group used primary neurons from the THY-Tau22 AD mouse model to show that tau affects the transport of tPA-containing vesicles in neurons [25][86]. However, further in vivo studies are necessary to better understand the role of tPA in tau pathology in AD.
Similar to tPA, uPA has been associated with plasmin-mediated clearance of Aβ in the brain [26][87]. It has also been suggested that uPA protects neurons from Aβ toxicity independently of plasmin by increasing the expression of cadherin [27][88]. The role of uPA in AD has been recently reviewed [27][88] and, therefore, will not be further discussed in there present review.
3. Plasminogen Activator Inhibitor-1 (PAI-1)
PAI-1 is a member of the serine protease inhibitor family. It plays a key role in the negative regulation of the fibrinolytic system by inhibiting the activity of tPA and uPA. PAI-1 also has been implicated in other functions, such as cell migration, angiogenesis, and wound healing [28][29][57,89]. Similar to plasminogen and tPA, PAI-1 is also reported to be involved in central nervous system physiology and pathology [30][31][90,91].
There is mounting evidence that suggests PAI-1 plays a role in the progression of AD. Elevated levels of PAI-1 have been observed in the brains of both AD mouse models and human patients, which correlates with an increase in Aβ load in both brain and the blood. For instance, in the APP/PS1 AD mouse line, PAI-1 levels increase over time as AD pathology worsens [30][90]. Moreover, PAI-1 deficiency in this mouse line then leads to reduced Aβ monomers, oligomers, and plaques in their brains [30][90]. Additionally, two independent studies using different PAI-1 inhibitors show a reduction in Aβ load and cognitive decline in AD mice [18][32][80,92]. Interestingly, these PAI-1 inhibitors targeted different types of Aβ, with TM5275 reducing both Aβ40 and Aβ42 load and PAI-039 only reducing Aβ40. The discrepancy in which Aβ form was reduced could be attributed to the use of different AD mouse models for these experiments, the nature of the PAI-1 inhibitors (i.e., different binding sites, efficacy, and off-target effects), or different timelines of PAI-1 inhibitor treatment.
The proposed mechanism by which PAI-1 inhibition leads to protection in AD is primarily associated with the increase in tPA and subsequent plasmin activity that results in the direct clearance of Aβ [18][30][32][80,90,92]. However, an increase in the maturation of BDNF after a decrease in PAI-1 inhibition may contribute to protection in AD [33][93], likely via an increase in plasmin and tPA activity. In contrast, an in vitro study using primary hippocampal neurons suggested that PAI-1 treatment may be neuroprotective in Aβ-mediated neurotoxicity [34][94]. However, to researcheours' knowledge, there are no in vivo studies further exploring this alternative hypothesis that PAI-1 could be neuroprotective in AD.
The findings from AD mouse model studies have led to increased interest in examining CSF or blood PAI-1 levels as a biomarker for AD in humans. One study found that PAI-1 plasma levels increase as AD progresses in humans [35][95]. Furthermore, in people with type 2 diabetes, a strong correlation was found between increased plasma PAI-1 levels in diabetics with MCI [36][96]. Another study found a positive correlation of PAI-1 levels with Aβ in the CSF of male but not female AD patients [37][97], indicating a potential sexual dimorphism of PAI-1 as an AD CSF biomarker. However, plasma PAI-1 levels were found to be increased in elderly, uneducated Chinese individuals with AD, but this finding was not observed in their educated AD counterparts [38][98]. These findings highlight the limitations of relying on a single biomarker to identify and diagnose AD, as disease populations are highly heterogeneous.
In terms of genetic screenings, a polymorphism in the PAI-1 promoter (4G/4G) that increases PAI-1 transcription has been linked to an increased risk for AD [39][99]. However, this was not observed in the Chinese Han population [40][100] nor in a recent meta-analysis [41][101]. It is important to note that there is one study showing that PAI-1 levels are unchanged in the brains of AD patients, although they show reduced tPA activity [20][82]. In this small study, neuroserpin, another inhibitor of tPA in the brain, was increased instead of PAI-1 [20][82]. This result suggests that not only PAI-1 but other inhibitors of tPA and plasmin may be involved in AD pathology. Further research is necessary to elucidate the involvement of other inhibitors of these proteins in AD pathogenesis.
4. Fibrinogen
Fibrinogen is a soluble plasma protein composed of three pairs of polypeptide chains—alpha, beta, and gamma—that is converted to an insoluble fibrin clot by the serine protease thrombin. Fibrinogen is secreted by platelets, hepatocytes, and endothelial cells in response to injury. After fibrinogen cleavage by thrombin, fibrin forms a mesh-like network that traps blood cells and other components to form a clot, preventing further bleeding and facilitating tissue repair. The cross-linking action of coagulation factor XIII then stabilizes the fibrin network. In several neurological disorders, including stroke and AD, fibrin(ogen) has been shown to extravasate into the brain [42][43][61,102]. There is substantial evidence showing that the fibrinogen beta chain binds to Aβ in vitro and in vivo, affecting clot formation, structure, and dissolution [44][45][103,104]. In AD mouse models, fibrin(ogen) deposits with Aβ in and around blood vessels, and its deposition in the brain parenchyma correlates with the progression of AD pathology, including neuronal death [46]. Numerous studies have shown that pharmacological or genetic reduction of fibrinogen reduces CAA pathology and/or parenchymal Aβ load and rescues behavioral deficits in various AD mouse lines [46][47][48][49][46,105,106,107]. The precise disruption of the Aβ/fibrinogen complex using the small molecule RU-505 leads to a reduction in CAA pathology, neuroinflammation, and cognitive decline in two AD mouse models [50][108], suggesting that fibrin(ogen) itself is not enough to induce AD pathology. Additionally, the interaction between fibrinogen and Aβ is proposed to enhance tPA-mediated plasminogen activation, which may contribute to CAA-associated intracerebral hemorrhage [51][52][33,109]. However, the Aβ/fibrinogen complex delays fibrin clot lysis [51][33]. This result suggests that the increase in plasmin activity may be ineffective in degrading Aβ/fibrinogen complexes and dissolving the clot, further contributing to CAA pathology. The molecular basis of how the fibrinolytic system directly participates in CAA and related pathologies requires further investigation.
Mechanistically, there is evidence that fibrinogen’s gamma chain is also involved in mediating AD pathology since targeting this region with a monoclonal antibody or an inhibitory peptide rescues neuropathology in AD mice [48][53][106,110]. This region is necessary for fibrinogen’s binding to the CD11b/Mac-1 (macrophage integrin) receptor, leading to microglial activation and promoting synaptic elimination in AD mice [54][111].
There is growing evidence linking fibrin(ogen) to AD pathology in humans. Increased fibrin(ogen) deposition has been observed in both blood vessels and the brain parenchyma of AD patients [47][54][105,111]. In addition, a recent study reported a positive correlation between plasma fibrinogen levels and Aβ and phosphorylated tau in the brains of AD patients [55][112]. However, two recent studies did not find a correlation between fibrinogen and AD risk or between brain deposition of fibrin(ogen) and Aβ or phosphorylated tau [56][57][113,114]. Therefore, further studies are needed to fully understand and clarify the role of fibrinogen in human AD pathology. Nonetheless, the use of plasma fibrinogen as an auxiliary biomarker in AD may be relevant for tailoring treatment strategies for individual patients.
A schematic that portrays vascular deficiencies involving the plasma contact system and the fibrinolytic system in AD can be found in Figure 1.
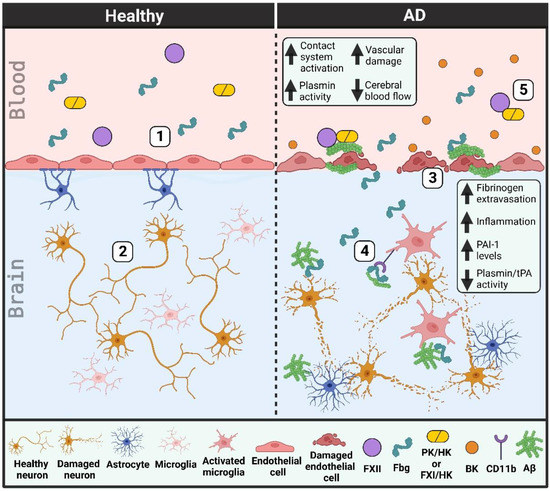
Figure 1. Vascular dysfunction in Alzheimer’s disease (AD) patient brains and blood vessels. (1) Healthy individuals have an intact blood-brain barrier. Endothelial cells are connected by tight junctions and lined by pericytes (not shown) and astrocytes. (2) This intact blood-brain barrier prevents the extravasation of blood proteins into the brain parenchyma, thereby helping to limit neuroinflammation and protect neurons from damage. (3) The blood-brain barrier in AD patients is damaged as tight junctions are lost and endothelial cells and pericytes (not depicted) degenerate. Cerebral blood flow is decreased due in part to an increase in beta-amyloid (Aβ) along blood vessel walls (cerebral amyloid angiopathy). (4) This vascular damage allows for the extravasation of blood proteins, like fibrinogen (Fbg), into the brain parenchyma. Fibrinogen may promote neuronal degeneration and induce an immune response in the brain, both through its interaction with Aβ and its binding to and activation of the CD11b/Mac-1 receptor on microglia. tPA and plasmin activities are decreased while PAI-1 levels are increased, limiting the removal of Aβ plaques from the brain parenchyma. (5) There is increased plasma contact system activation in AD patient blood due to enhanced interaction of plasma Aβ with factor XII (FXII). Bradykinin (BK) levels are increased, promoting neuroinflammation and further vascular damage. (PK, plasma kallikrein; FXI, factor XI; HK, high molecular weight kininogen).