The coronavirus 2019 (COVID-19) pandemic has caused a huge impact on health and economic issues. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) causes cellular damage by entry mediated by the angiotensin-converting enzyme 2 of the host cells and its conjugation with spike proteins of SARS-CoV-2. Beyond airway infection and acute respiratory distress syndrome, acute kidney injury is common in SARS-CoV-2-associated infection, and acute kidney injury (AKI) is predictive to multiorgan dysfunction in SARS-CoV-2 infection. Beyond the cytokine storm and hemodynamic instability, SARS-CoV-2 might directly induce kidney injury and cause histopathologic characteristics, including acute tubular necrosis, podocytopathy and microangiopathy. The expression of apparatus mediating SARS-CoV-2 entry, including angiotensin-converting enzyme 2, transmembrane protease serine 2 (TMPRSS2) and a disintegrin and metalloprotease 17 (ADAM17), within the renal tubular cells is highly associated with acute kidney injury mediated by SARS-CoV-2. Both entry from the luminal and basolateral sides of the renal tubular cells are the possible routes for COVID-19, and the microthrombi associated with severe sepsis and the dysregulated renin–angiotensin–aldosterone system worsen further renal injury in SARS-CoV-2-associated AKI. In the podocytes of the glomerulus, injured podocyte expressed CD147, which mediated the entry of SARS-CoV-2 and worsen further foot process effacement, which would worsen proteinuria, and the chronic hazard induced by SARS-CoV-2-mediated kidney injury is still unknown. Therefore, the aim of the review is to summarize current evidence on SARS-CoV-2-associated AKI and the possible pathogenesis directly by SARS-CoV-2.
- COVID-19
- acute kidney injury
- ACE2
- ADAM17
- TMPRSS2
- CD147
1. Introduction
The coronavirus disease 2019 (COVID-19) pandemic has had severe public health and economic impacts as a result of its rapid spread and association with severe acute respiratory distress syndrome. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) can lead to severe sepsis and systemic inflammation, which can induce multiple organ dysfunction. Acute kidney injury (AKI) is predictive of mortality and deteriorating organ dysfunction in patients with SARS-CoV-2 [1,2][1][2]. The most common risk factors related to SARS-Cov-2-mediated AKI include diabetes, obesity or hypertension and previous chronic kidney disease (CKD) [3]. Angiotensin-converting enzyme 2 (ACE2) and an assembly protein are known requirements for the entry of the virus into host cells. In patients with hypertension, diabetes mellitus and CKD, renin–angiotensin–aldosterone system activation is associated with ACE2 deficiency, which worsens renal tubulointerstitial fibrosis [4]. The modified phenotype of podocyte might facilitate SARS-CoV-2 entry into the reno-tubular system and worsen kidney damage. Moreover, SARS-CoV-2-associated sepsis might worsen AKI through the induction of a cytokine storm or hemodynamic dysregulation [5], but the pathogenesis and understanding of SARS-CoV-2-mediated kidney injury is still lacking. This review investigated the possible molecular mechanisms of acute kidney injury directly mediated by SARS-CoV-2.
2. High Levels of Angiotensin-Converting Enzyme 2 and Transmembrane Protease Serine 2 Expression in the Urinary System
According to data published by the Genotype-Tissue Expression project, ACE2 is highly expressed within the kidneys [6]. ACE2 is distributed throughout multiple cells within the kidneys and is most concentrated in proximal tubule cells [7]. Fan et al. suggested that specific ACE2 expression is high within proximal tubular cells but is not observed in immune cells or glomerular parietal epithelial cells [8]. Suryawanshi et al. analyzed the data of kidney tissues in single-cell RNA sequencing datasets and found that proximal tubular cells in the kidney co-express ACE2 and transmembrane protease serine 2 (TMPRSS2) [9]. Studies have reported high levels of ACE2 and TMPRSS2 expression in proximal renal tubular epithelial cells and kidney podocytes [10–12][10][11][12], and such characteristics facilitate the SARS-CoV replication within these cells [13].
3. TMPRSS2 and a Disintegrin and Metalloprotease 17 (ADAM17) Protect Against Viruses in Host Proteases
ACE2 within the cell membrane is important for SARS-CoV-2 entry into host cells [14]. However, in severe acute respiratory syndrome coronavirus 1 (SARS-CoV-1) or SARS-CoV-2 infection, poor prognosis is reportedly associated with ACE2 downregulation [15]. Theoretically, fewer viral entry routes should correspond to improved clinical outcomes. However, studies have demonstrated that decreased ACE2 expression levels lead to greater illness severity and more serious end-organ damage because ACE2 plays an anti-inflammatory role under renin–angiotensin–aldosterone system (RAAS) activation [16]. Classical RAAS involves the conversion of Angiotensin (Ang) I to Ang II and AngII binding with angiotensin-1 receptor (AT1R). The classic RAAS pathway involves vasoconstriction, oxidative stress, inflammation and fibrosis. The non-classical RAAS pathway, on the other hand, converts AngII to Ang(1–7) by ACE2, and the binding of Ang(1–7) with Mas receptor (MasR) provides vasodilatation and an anti-inflammatory effect [17]. Viral invasion into host cells occurs primarily through the ACE2 receptor; this receptor may mediate the entry of SARS-CoV-2 into host cells through two distinct routes. The first involves clathrin-dependent endocytosis and the second involves ACE2 receptor–mediated TMPRSS2-dependent membrane fusion [18].
ACE2 downregulation occurs as these proteins are shed from cell membranes and circulated throughout the body. The end product of ACE2 cleavage mediated by a disintegrin and metalloprotease 17 (ADAM17) and TMPRSS2 may play a protective role against SARS-CoV-2 entry. In SARS-CoV-2, this occurs as a result of the spike protein activating ACE2 expression by seizing two host proteases: TMPRSS2, which facilitates viral entry by cleaving the S antigen into S1 (the active binding site), and ADAM17, which downregulates ACE2 expression by shedding ACE2 proteins into soluble form. The soluble ACE2 directly attached virus within circulation and decreased SARS-CoV-2 entry [19,20][19][20]. Research has indicated that ACE2 within human embryonic kidney cells can be shed by ADAM17 and protect against SARS-S entry [21]. In the human embryonic kidney cell line, decreased ACE2 expression is responsible for SARS-CoV-2 complications and end-organ damage and may cause greater harm to the host through enhanced ACE2 toxic effects, such as the activation of proinflammatory cytokines [22]. Heurich et al. suggested that TMPRSS2 and ADAM17 participate in the differential internalization of SARS-S [23]. TMPRSS2 cleaves ACE2 in arginine and lysine residues within amino acids 697 to 716, and the cleaved ACE2 is essential for SARS-S entry. Entry through TMPRSS2 is unrelated to ADAM17 because TRPMSS2 competes with ADAM17. Therefore, ADAM17 was proposed to participate in ACE2 ectodomain shedding, and TMPRSS2 enhances the intracellular cleavage of ACE2; thus, ADAM17 and TMPRSS2 compete during ACE2 processing. Hoffmann et al. demonstrated that TMPRSS2 inhibitors can block SARS-S cell entry through S protein processing [24]. Research has indicated that recombinant human ACE2 (rhACE2) mimics soluble ACE2 and inhibits viral invasion into host cells through competitive binding to CoV with the ACE2 lying in cellular membrane. The circulatory rhACE2-CoV2 binding decreases the internalization of membranous ACE2. Therefore, the non-classical RAAS provides further protection by alleviating intracellular inflammation [25]. Monteil et al. also used rhACE2 to block SARS-CoV-2 entry into the kidney organoid, demonstrating the possible protective effect of cleaved ACE2 against COVID-19 entry into host cells [26]. Therefore, in addition to its absolute expression, the cleavage and shedding of ACE2 could influence COVID-19 entry into cells ((Figure 1).
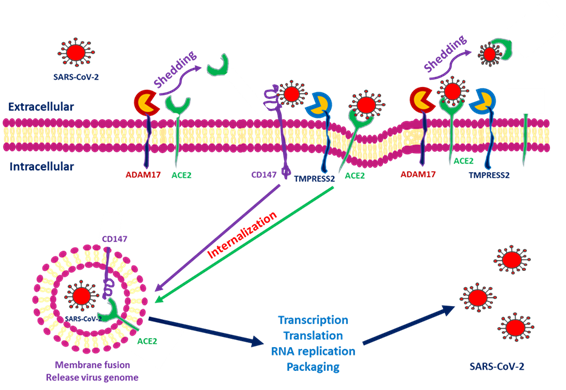
Figure 1. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) entry into cells through interactions among angiotensin-converting enzyme 2 (ACE2), transmembrane protease serine 2 (TMPRSS2), a disintegrin and metalloprotease 17 (ADAM17) and CD147. After SARS-CoV-2 interacts with the cells, ACE2 conjugates with the spike protein of SARS-CoV-2. TMPRSS2 interacts with the spike protein and facilitates internalization. ADAM17 serves to shed SARS-CoV-2 by transforming ACE2 into a soluble form. CD147 expressed within the transmembrane also facilitates entry and induces downstream inflammatory cytokine expression.
However, more studies are necessary to clarify the role of ADAM17 and other proteases on ACE2 shedding in the kidney and the importance of these proteases in SARS-CoV-2 infection [27].
4. SARS-CoV-2 Invades Host Cells Through a Novel Route: CD147-Spike Protein
Research on kidney epithelial cells (Vero E6 cells) has demonstrated that spike proteins bound to CD147 can mediate SARS-CoV-2 host invasion. The localization of CD147 and spike protein was observed in SARS-CoV-2–infected cells through an immunoelectron microscope, and the discovery of the new CD147-SP route for SARS-CoV-2 invasion of host cells has provided a critical target for the development of a specific antiviral medicine [28].
CD147 (Extracellular Matrix Metalloproteinase Inducer/Basigin) in Kidney Diseases
Basigin (Bsg)/CD147, also known as an extracellular matrix metalloproteinase (MMP) inducer (EMMPRIN), is the glycosylated transmembrane protein governing cell survival, cell migration and cancer invasion [29]. In SARS-CoV-2–mediated myocardial injury, EMMPRIN facilitates viral entry into the cardiomyocyte and induces the release of cytokines, such as interleukin-18 [30]. Moreover, the reactive oxygen species generated during inflammation dysregulate myocardial remodeling [31]. Atrial fibrillation is more inducible after SARS-CoV-2 entry and the downstream release of cytokines [32]. Wang et al. reported that the high affinity of CD147 and the spike protein to SARS-CoV-2 mediates viral entry into the kidney epithelium [28]. In normal kidneys, CD147 is highly expressed only on the basolateral side of tubular epithelial cells [29]. In the case of viremia, SARS-CoV-2 might invade renal proximal tubular epithelial cells through both the luminal surface and the basolateral side (Figure 2). Kato et al. demonstrated that Bsg/CD147 was associated with neutrophil recruitment in renal tubules with ischemic/reperfusion injury. In Bsg-deficient [Bsg(-/-)] mice, neutrophilic infiltration is suppressed after renal ischemic/reperfusion injury. Bsg/CD147 is a crucial physiologic ligand for E-selectin and therefore facilitates neutrophilic infiltration [33]. In an animal model of unilateral urinary obstruction, Bsg/CD147 promoted renal fibrosis by inducing MMP and hyaluronan expression. In a primary culture of the renal tubules of Bsg(-/-) mice, MMP expression was less responsive to transforming growth factor β [34].
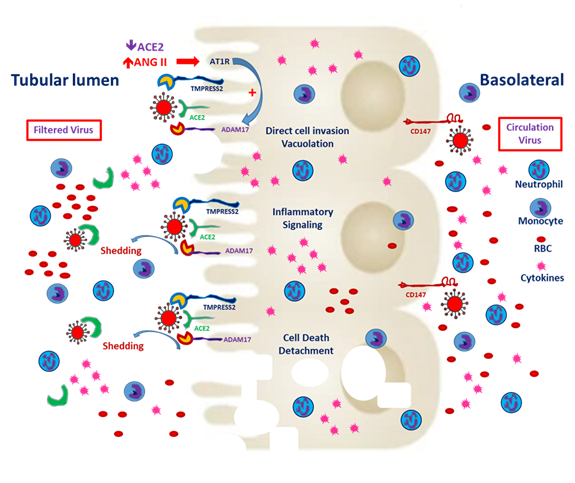
Figure 2. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) entry from the apical and basolateral sides of proximal tubular cells. CD147 expression is mainly distributed on the basolateral side of the proximal tubules. In the tubules predisposed to injury, enhanced CD147 might facilitate the entry of SARS-CoV-2. Classical RAAS activation is common in CKD patients, and local expression of angiotensin-converting enzyme 2 (ACE2) enhances hypercoagulability and induces microthrombi. Besides, filtered SARS-CoV-2 might enter the proximal tubules through ACE2 and TMPRESS2. Entry through either route could worsen tubular inflammation and increase cell death.
The aforementioned research findings suggest that SARS-CoV-2 targets the renal tubules by entering from the basolateral side in adjunction with CD147. Yoshioka et al. reported that Bsg/CD147 is rarely expressed in healthy podocytes. In adriamycin-induced nephropathy, increases in Bsg expression and proteinuria are mediated by increases in focal adhesion kinase signaling [35]. In their clinical study, Musial et al. reported the serum concentrations of CD147 along with other fibrosis markers, such as transforming growth factor beta (TGF-β) levels, in children with CKD [36]. The findings indicated that patients with CKD may exhibit variations in CD147 expression that may contribute to pathological changes.
5. COVID-19 Prevents ACE2 from Converting ANG II into ANG I–VII and Increases Intracellular ANG II and Membrane ADAM17 Expression
Cytosolic ACE2 is a negative regulator for the activation of ACE and Ang I [37]. The carboxypeptidase ACE2 converts Ang II into Ang (1–7) and Ang I into Ang (1–9), thus preventing the conversion of Ang I into Ang II [38]. Under normal conditions, endothelial cells synthesize tissue factors and inhibitors of thrombosis to maintain immune and coagulation homeostasis [39]. COVID-19 enters cells through ACE2-mediated assembly. The inability of ACE2 to convert Ang I or Ang II when paired with COVID-19 results in endothelial senescence, caused by the upregulation of interleukin 6 and oxidative stress induced by disturbances in mitochondrial function [40]. In the hypoxia and sepsis, the un-countered AT1R activation by Ang II worsens the vasoconstriction and downstream fibrosis within lung tissue [37]. The excess Ang I and II induces immune and coagulation abnormalities and dysfunction. COVID-19/ACE2 complexes enter host cells through endocytosis, and those that are not endocytosed are shed by ADAM17 [40]. Increases in renal ADAM17 expression are known to mediate Ang II-induced growth of renal lesions in patients with CKD. The binding of Ang II to its AT1 receptor increases cytosolic ADAM17 expression and its translocation to the plasma membrane. In the vascular system, excessive ADAM17 expression induces the shedding of ACE2 and worsens further activation of the classical RAAS and inflammation [41]. ADAM17 induces the shedding of ACE2 and increases circulatory ACE2 (sACE2), thereby mitigating further entry of COVID-19 [42]. Additionally, ADAM17 modulates the shedding of transforming growth factor-α (TGF-α) from its transmembrane precursor and activates the epidermal growth factor receptor (EGFR), causing proteinuria, tubular hyperplasia, fibrosis and mononuclear cell infiltration [43]. TGF-α/EGFR-driven vitamin D receptor reduction impairs 1,25(OH)2D/VDR renoprotection [44]. ADAM17 is also expressed within the renal tubules, and its activation, mediated by classical RAAS activation, may prevent the COVID-19 shed by sACE2 from entering tubular cells [45]. However, this further activation of the classical RAAS may worsen the hypercoagulability of COVID-19 infection, thereby increasing the extent of tubular damage and the risk of damage to the interstitium.
References
- Chen, Y.-T.; Shao, S.-C.; Lai, E. C.-C.; Hung, M.-J.; Chen, Y.-C., Mortality rate of acute kidney injury in SARS, MERS, and COVID-19 infection: a systematic review and meta-analysis. Crit Care 2020, 24, (1), 439-439.
- Cheng, Y.; Wang, W.; Wu, L.; Cai, G., SARS-CoV-2-Related Kidney Injury: Current Concern and Challenges. SN Comprehensive Clinical Medicine 2020.
- Nadim, M. K.; Forni, L. G.; Mehta, R. L.; Connor, M. J.; Liu, K. D.; Ostermann, M.; Rimmelé, T.; Zarbock, A.; Bell, S.; Bihorac, A.; Cantaluppi, V.; Hoste, E.; Husain-Syed, F.; Germain, M. J.; Goldstein, S. L.; Gupta, S.; Joannidis, M.; Kashani, K.; Koyner, J. L.; Legrand, M.; Lumlertgul, N.; Mohan, S.; Pannu, N.; Peng, Z.; Perez-Fernandez, X. L.; Pickkers, P.; Prowle, J.; Reis, T.; Srisawat, N.; Tolwani, A.; Vijayan, A.; Villa, G.; Yang, L.; Ronco, C.; Kellum, J. A., COVID-19-associated acute kidney injury: consensus report of the 25th Acute Disease Quality Initiative (ADQI) Workgroup. Nature Reviews Nephrology 2020.
- Nangaku, M.; Fujita, T., Activation of the renin-angiotensin system and chronic hypoxia of the kidney. Hypertens Res 2008, 31, (2), 175-84.
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; Cheng, Z.; Yu, T.; Xia, J.; Wei, Y.; Wu, W.; Xie, X.; Yin, W.; Li, H.; Liu, M.; Xiao, Y.; Gao, H.; Guo, L.; Xie, J.; Wang, G.; Jiang, R.; Gao, Z.; Jin, Q.; Wang, J.; Cao, B., Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. The Lancet 2020, 395, (10223), 497-506.
- Chen, L.; Li, X.; Chen, M.; Feng, Y.; Xiong, C., The ACE2 expression in human heart indicates new potential mechanism of heart injury among patients infected with SARS-CoV-2. Cardiovascular Research 2020, 116, (6), 1097-1100.
- Zou, X.; Chen, K.; Zou, J.; Han, P.; Hao, J.; Han, Z., Single-cell RNA-seq data analysis on the receptor ACE2 expression reveals the potential risk of different human organs vulnerable to 2019-nCoV infection. Frontiers of Medicine 2020, 14, (2), 185-192.
- Fan, C.; Li, K.; Ding, Y.; Lu, W. L.; Wang, J., ACE2 Expression in Kidney and Testis May Cause Kidney and Testis Damage After 2019-nCoV Infection. medRxiv 2020, 2020.02.12.20022418.
- Suryawanshi, H.; Morozov, P.; Muthukumar, T.; tenOever, B. R.; Yamaji, M.; Williams, Z.; Tuschl, T., Cell-Type-Specific Expression of Renin-Angiotensin-System Components in the Human Body and Its Relevance to SARS-CoV-2 Infection. bioRxiv 2020, 2020.04.11.034603.
- Pan, X.-w.; Xu, D.; Zhang, H.; Zhou, W.; Wang, L.-h.; Cui, X.-g., Identification of a potential mechanism of acute kidney injury during the COVID-19 outbreak: a study based on single-cell transcriptome analysis. Intensive Care Medicine 2020, 46, (6), 1114-1116.
- Dong, M.; Zhang, J.; Ma, X.; Tan, J.; Chen, L.; Liu, S.; Xin, Y.; Zhuang, L., ACE2, TMPRSS2 distribution and extrapulmonary organ injury in patients with COVID-19. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie 2020, 131, 110678.
- Qi, J.; Zhou, Y.; Hua, J.; Zhang, L.; Bian, J.; Liu, B.; Zhao, Z.; Jin, S., The scRNA-seq expression profiling of the receptor ACE2 and the cellular protease TMPRSS2 reveals human organs susceptible to COVID-19 infection. bioRxiv 2020, 2020.04.16.045690.
- Pacciarini, F.; Ghezzi, S.; Canducci, F.; Sims, A.; Sampaolo, M.; Ferioli, E.; Clementi, M.; Poli, G.; Conaldi, P. G.; Baric, R.; Vicenzi, E., Persistent replication of severe acute respiratory syndrome coronavirus in human tubular kidney cells selects for adaptive mutations in the membrane protein. J Virol 2008, 82, (11), 5137-5144.
- Kai, H.; Kai, M., Interactions of coronaviruses with ACE2, angiotensin II, and RAS inhibitors—lessons from available evidence and insights into COVID-19. Hypertension Research 2020, 43, (7), 648-654.
- Du, F.; Liu, B., COVID-19: the role of excessive cytokine release and potential ACE2 down-regulation in promoting hypercoagulable state associated with severe illness. 2020, 1-17.
- AlGhatrif, M.; Cingolani, O.; Lakatta, E. G., The Dilemma of Coronavirus Disease 2019, Aging, and Cardiovascular Disease: Insights From Cardiovascular Aging Science. JAMA Cardiology 2020, 5, (7), 747-748.
- Chappell, M. C., Nonclassical renin-angiotensin system and renal function. Compr Physiol 2012, 2, (4), 2733-2752.
- Kuba, K.; Imai, Y.; Ohto-Nakanishi, T.; Penninger, J. M., Trilogy of ACE2: a peptidase in the renin-angiotensin system, a SARS receptor, and a partner for amino acid transporters. Pharmacol Ther 2010, 128, (1), 119-128.
- Brill, A.; Chauhan, A. K.; Canault, M.; Walsh, M. T.; Bergmeier, W.; Wagner, D. D., Oxidative stress activates ADAM17/TACE and induces its target receptor shedding in platelets in a p38-dependent fashion. Cardiovasc Res 2009, 84, (1), 137-44.
- Jia, H. P.; Look, D. C.; Tan, P.; Shi, L.; Hickey, M.; Gakhar, L.; Chappell, M. C.; Wohlford-Lenane, C.; Paul B. McCray, J., Ectodomain shedding of angiotensin converting enzyme 2 in human airway epithelia. American Journal of Physiology-Lung Cellular and Molecular Physiology 2009, 297, (1), L84-L96.
- Lambert, D. W.; Yarski, M.; Warner, F. J.; Thornhill, P.; Parkin, E. T.; Smith, A. I.; Hooper, N. M.; Turner, A. J., Tumor necrosis factor-alpha convertase (ADAM17) mediates regulated ectodomain shedding of the severe-acute respiratory syndrome-coronavirus (SARS-CoV) receptor, angiotensin-converting enzyme-2 (ACE2). The Journal of biological chemistry 2005, 280, (34), 30113-9.
- Luther, J. M.; Gainer, J. V.; Murphey, L. J.; Yu, C.; Vaughan, D. E.; Morrow, J. D.; Brown, N. J., Angiotensin II induces interleukin-6 in humans through a mineralocorticoid receptor-dependent mechanism. Hypertension (Dallas, Tex. : 1979) 2006, 48, (6), 1050-7.
- Heurich, A.; Hofmann-Winkler, H.; Gierer, S.; Liepold, T.; Jahn, O.; Pöhlmann, S., TMPRSS2 and ADAM17 cleave ACE2 differentially and only proteolysis by TMPRSS2 augments entry driven by the severe acute respiratory syndrome coronavirus spike protein. J Virol 2014, 88, (2), 1293-1307.
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T. S.; Herrler, G.; Wu, N. H.; Nitsche, A.; Müller, M. A.; Drosten, C.; Pöhlmann, S., SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, (2), 271-280.e8.
- Wu, J.; Deng, W.; Li, S.; Yang, X., Advances in research on ACE2 as a receptor for 2019-nCoV. 2020, 1-14.
- Monteil, V.; Kwon, H.; Prado, P.; Hagelkrüys, A.; Wimmer, R. A.; Stahl, M.; Leopoldi, A.; Garreta, E.; Hurtado Del Pozo, C.; Prosper, F.; Romero, J. P.; Wirnsberger, G.; Zhang, H.; Slutsky, A. S.; Conder, R.; Montserrat, N.; Mirazimi, A.; Penninger, J. M., Inhibition of SARS-CoV-2 Infections in Engineered Human Tissues Using Clinical-Grade Soluble Human ACE2. Cell 2020, 181, (4), 905-913.e7.
- Palau, V.; Riera, M.; Soler, M. J., ADAM17 inhibition may exert a protective effect on COVID-19. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association 2020, 35, (6), 1071-1072.
- Wang, K.; Chen, W.; Zhou, Y.-S.; Lian, J.-Q.; Zhang, Z.; Du, P.; Gong, L.; Zhang, Y.; Cui, H.-Y.; Geng, J.-J.; Wang, B.; Sun, X.-X.; Wang, C.-F.; Yang, X.; Lin, P.; Deng, Y.-Q.; Wei, D.; Yang, X.-M.; Zhu, Y.-M.; Zhang, K.; Zheng, Z.-H.; Miao, J.-L.; Guo, T.; Shi, Y.; Zhang, J.; Fu, L.; Wang, Q.-Y.; Bian, H.; Zhu, P.; Chen, Z.-N., SARS-CoV-2 invades host cells via a novel route: CD147-spike protein. bioRxiv 2020, 2020.03.14.988345.
- Kosugi, T.; Maeda, K.; Sato, W.; Maruyama, S.; Kadomatsu, K., CD147 (EMMPRIN/Basigin) in kidney diseases: from an inflammation and immune system viewpoint. Nephrology Dialysis Transplantation 2014, 30, (7), 1097-1103.
- Venkatesan, B.; Valente, A. J.; Prabhu, S. D.; Shanmugam, P.; Delafontaine, P.; Chandrasekar, B., EMMPRIN activates multiple transcription factors in cardiomyocytes, and induces interleukin-18 expression via Rac1-dependent PI3K/Akt/IKK/NF-kappaB andMKK7/JNK/AP-1 signaling. J Mol Cell Cardiol 2010, 49, (4), 655-63.
- Cao, M.; Yuan, W.; Peng, M.; Mao, Z.; Zhao, Q.; Sun, X.; Yan, J., Role of CyPA in cardiac hypertrophy and remodeling. Bioscience reports 2019, 39, (12), BSR20193190.
- Gawałko, M.; Kapłon-Cieślicka, A.; Hohl, M.; Dobrev, D.; Linz, D., COVID-19 associated atrial fibrillation: Incidence, putative mechanisms and potential clinical implications. IJC Heart & Vasculature 2020, 30, 100631.
- Kato, N.; Yuzawa, Y.; Kosugi, T.; Hobo, A.; Sato, W.; Miwa, Y.; Sakamoto, K.; Matsuo, S.; Kadomatsu, K., The E-selectin ligand basigin/CD147 is responsible for neutrophil recruitment in renal ischemia/reperfusion. J Am Soc Nephrol 2009, 20, (7), 1565-76.
- Kato, N.; Kosugi, T.; Sato, W.; Ishimoto, T.; Kojima, H.; Sato, Y.; Sakamoto, K.; Maruyama, S.; Yuzawa, Y.; Matsuo, S.; Kadomatsu, K., Basigin/CD147 promotes renal fibrosis after unilateral ureteral obstruction. The American journal of pathology 2011, 178, (2), 572-9.
- Yoshioka, T.; Kosugi, T.; Masuda, T.; Watanabe, T.; Ryuge, A.; Nagaya, H.; Maeda, K.; Sato, Y.; Katsuno, T.; Kato, N.; Ishimoto, T.; Yuzawa, Y.; Maruyama, S.; Kadomatsu, K., CD147/Basigin Deficiency Prevents the Development of Podocyte Injury through FAK Signaling. The American journal of pathology 2019, 189, (7), 1338-1350.
- Musiał, K.; Bargenda, A.; Zwolińska, D., Urine matrix metalloproteinases and their extracellular inducer EMMPRIN in children with chronic kidney disease. Renal failure 2015, 37, (6), 980-4.
- Alfano, G.; Guaraldi, G.; Fontana, F.; Ferrari, A.; Magistroni, R.; Mussini, C.; Cappelli, G., The Role of the Renin-Angiotensin System in Severe Acute Respiratory Syndrome-CoV-2 Infection. Blood Purif 2020, 1-5.
- Danser, A. H. J.; Epstein, M.; Batlle, D., Renin-Angiotensin System Blockers and the COVID-19 Pandemic: At Present There Is No Evidence to Abandon Renin-Angiotensin System Blockers. Hypertension (Dallas, Tex. : 1979) 2020, 75, (6), 1382-1385.
- Tang, N.; Li, D.; Wang, X.; Sun, Z., Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. Journal of Thrombosis and Haemostasis 2020, 18, (4), 844-847.
- Sfera, A.; Osorio, C.; Jafri, N.; Diaz, E. L.; Campo Maldonado, J. E., Intoxication With Endogenous Angiotensin II: A COVID-19 Hypothesis. Frontiers in Immunology 2020, 11, (1472).
- de Queiroz, T. M.; Lakkappa, N.; Lazartigues, E., ADAM17-Mediated Shedding of Inflammatory Cytokines in Hypertension. Frontiers in pharmacology 2020, 11, 1154.
- Xu, J.; Sriramula, S.; Xia, H.; Moreno-Walton, L.; Culicchia, F.; Domenig, O.; Poglitsch, M.; Lazartigues, E., Clinical Relevance and Role of Neuronal AT(1) Receptors in ADAM17-Mediated ACE2 Shedding in Neurogenic Hypertension. Circulation research 2017, 121, (1), 43-55.
- Melenhorst, W. B.; Visser, L.; Timmer, A.; van den Heuvel, M. C.; Stegeman, C. A.; van Goor, H., ADAM17 upregulation in human renal disease: a role in modulating TGF-alpha availability? American journal of physiology. Renal physiology 2009, 297, (3), F781-90.
- Morgado-Pascual, J. L.; Rayego-Mateos, S.; Valdivielso, J. M.; Ortiz, A.; Egido, J.; Ruiz-Ortega, M., Paricalcitol Inhibits Aldosterone-Induced Proinflammatory Factors by Modulating Epidermal Growth Factor Receptor Pathway in Cultured Tubular Epithelial Cells. BioMed research international 2015, 2015, 783538.
- Kato, T.; Hagiyama, M.; Ito, A., Renal ADAM10 and 17: Their Physiological and Medical Meanings. Frontiers in cell and developmental biology 2018, 6, 153.