Macrophages are cells in the innate immune system that provide the first line of defense against invading pathogens. Macrophages are classified broadly within two types of polarization states: classically activated (M1) and alternatively activated (M2). Hydrogen sulfide (H2S) has been increasingly recognized as a crucial inflammatory mediator in immune cells, particularly macrophages, due to its direct and indirect effects on cellular signaling, redox homeostasis, and energy metabolism. The intricate regulation of endogenous H2S production and metabolism involves the coordination of transsulfuration pathway (TSP) enzymes and sulfide oxidizing enzymes, with TSP’s role at the intersection of the methionine pathway and glutathione synthesis reactions.
- hydrogen sulfide
- macrophage
- inflammation
- cellular metabolism
1. Introduction
2. Hydrogen Sulfide and Redox Homeostasis
3. The Therapeutic Potential of Hydrogen Sulfide and Polysulfides
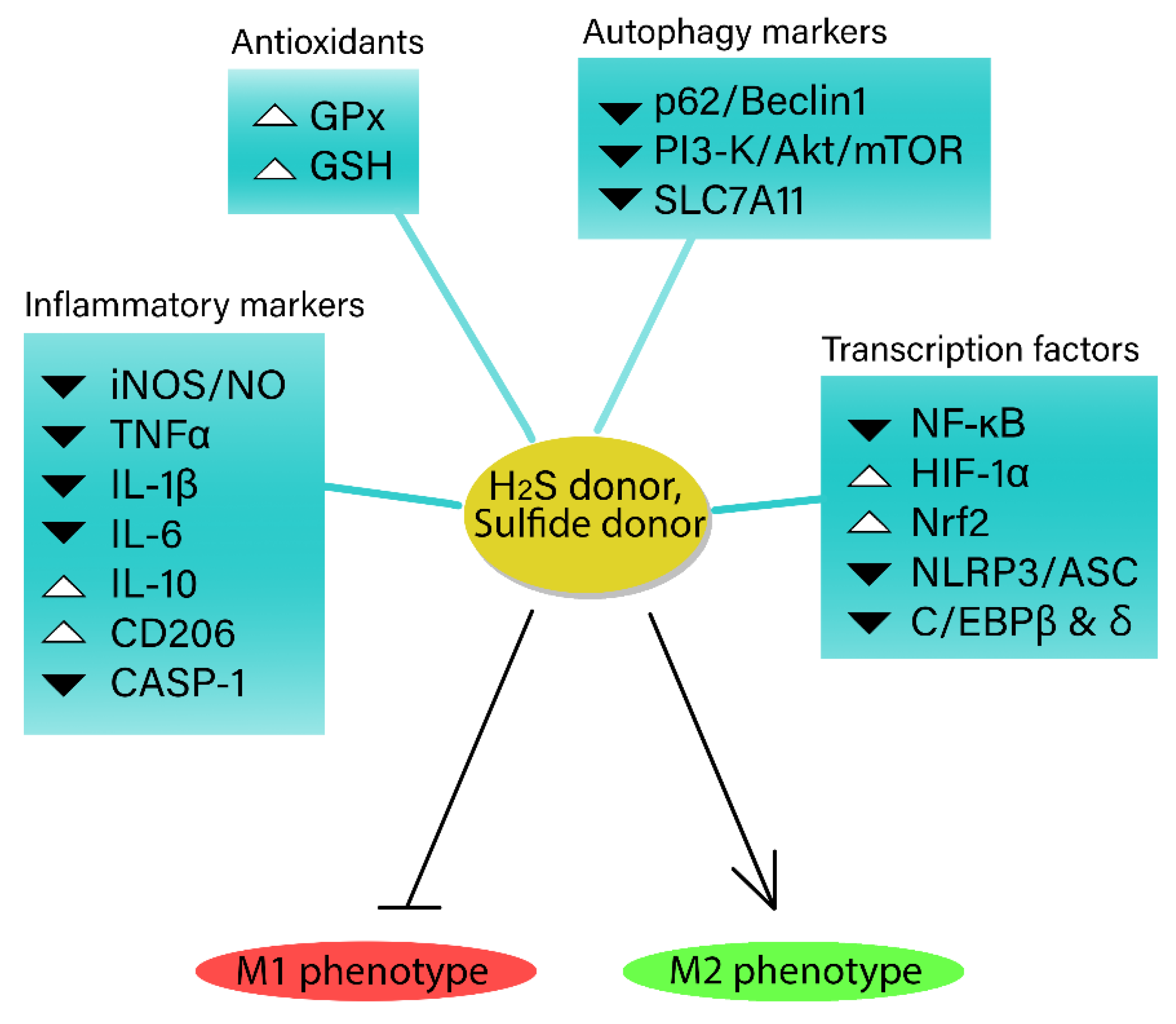
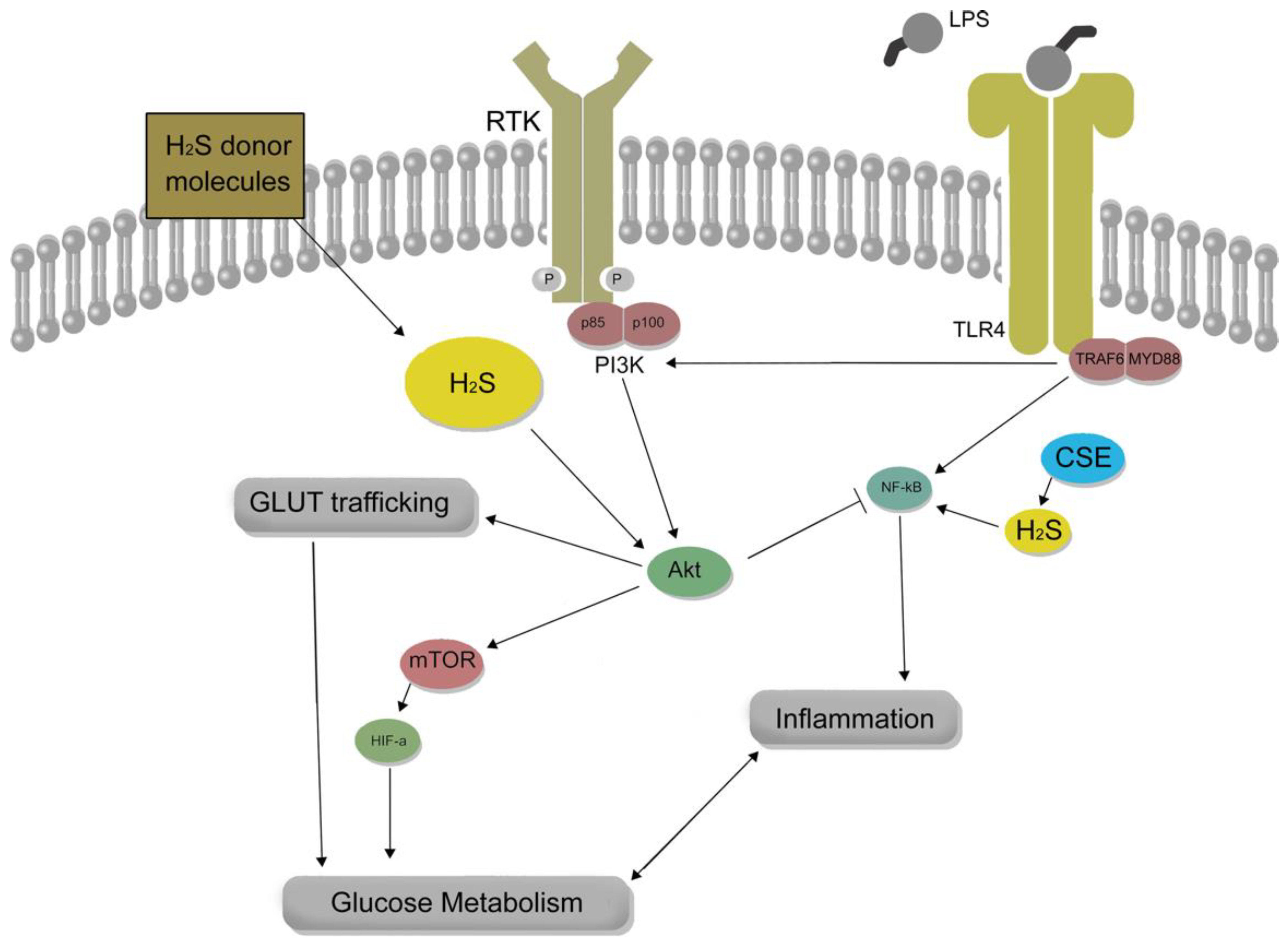
4. The Complex Role of Hydrogen Sulfide in Modulating Cellular Energy Metabolism and Inflammatory Responses in Macrophages
The regulatory function of H2S in mitochondrial respiration has become a focal point of discussion, owing to its potential to modulate cellular bioenergetics. Despite the uncertainty surrounding the effects of H2S on the inflammatory responses of immune cells, recent investigations have illuminated the association between H2S and cellular energy metabolism. H2S demonstrates a biphasic impact on cellular energy metabolism and may be implicated in the reconfiguration of inflammatory metabolism. This elaborate interplay underscores the capacity of H2S to influence cellular energy metabolism and impose heightened glucose substrate demands on cells exposed to H2S. Nevertheless, this relationship remains poorly understood, necessitating additional research to unravel the specific mechanisms underpinning this intricate association and the consequences of these effects in various pathological conditions. The interaction between exogenous and endogenous H2S and glucose metabolism presents a captivating area of exploration. H2S seemingly fosters a favorable redox environment for glycolysis, alluding to the possibility of further crosstalk. Intriguingly, H2S has also been found to induce alterations in transcription factors and proteins, bolstering the expression of inflammatory Glut1 in macrophages. The present review emphasizes the importance of continued research to disentangle the complex interrelations between H2S and cellular energy metabolism, ultimately unveiling novel insights into the broader implications of this signaling molecule.
References
- Herbst, S.; Schaible, U.E.; Schneider, B.E. Interferon Gamma Activated Macrophages Kill Mycobacteria by Nitric Oxide Induced Apoptosis. PLoS ONE 2011, 6, e19105.
- Piedrafita, D.; Parsons, J.C.; Sandeman, R.M.; Wood, P.R.; Estuningsih, S.E.; Partoutomo, S.; Spithill, T.W. Antibody-Dependent Cell-Mediated Cytotoxicity to Newly Excysted Juvenile Fasciola Hepatica in Vitro Is Mediated by Reactive Nitrogen Intermediates. Parasite Immunol. 2001, 23, 473–482.
- Thomas, G.R.; McCrossan, M.; Selkirk, M.E. Cytostatic and Cytotoxic Effects of Activated Macrophages and Nitric Oxide Donors on Brugia Malayi. Infect. Immun. 1997, 65, 2732–2739.
- Lavin, Y.; Mortha, A.; Rahman, A.; Merad, M. Regulation of Macrophage Development and Function in Peripheral Tissues. Nat. Rev. Immunol. 2015, 15, 731–744.
- Sun, H.-J.; Wu, Z.-Y.; Nie, X.-W.; Bian, J.-S. Role of Hydrogen Sulfide and Polysulfides in Neurological Diseases: Focus on Protein S-Persulfidation. Curr. Neuropharmacol. 2021, 19, 868.
- Wang, X.-Q.; Congyi, W.; Sun, F.; Luo, J.; Yue, T.; Wang, F.; Yang, C.; Zhang, S. The Hydrogen Sulfide Signaling in Macrophages: A Foe or Friend? Authorea Prepr. 2020.
- Bhatia, M.; Gaddam, R.R. Hydrogen Sulfide in Inflammation: A Novel Mediator and Therapeutic Target. Antioxid Redox Signal 2021, 34, 1368–1377.
- Fagone, P.; Mazzon, E.; Bramanti, P.; Bendtzen, K.; Nicoletti, F. Gasotransmitters and the Immune System: Mode of Action and Novel Therapeutic Targets. Eur. J. Pharmacol. 2018, 834, 92–102.
- Palmieri, E.M.; McGinity, C.; Wink, D.A.; McVicar, D.W. Nitric Oxide in Macrophage Immunometabolism: Hiding in Plain Sight. Metabolites 2020, 10, 429.
- Morse, D.; Choi, A.M.K. Heme Oxygenase-1: The “Emerging Molecule” Has Arrived. Am. J. Respir. Cell Mol. Biol. 2002, 27, 8–16.
- Paul, B.D.; Snyder, S.H. Gasotransmitter Hydrogen Sulfide Signaling in Neuronal Health and Disease. Biochem. Pharmacol. 2018, 149, 101–109.
- McBean, G.J.; Aslan, M.; Griffiths, H.R.; Torrão, R.C. Thiol Redox Homeostasis in Neurodegenerative Disease. Redox Biol. 2015, 5, 186–194.
- Sbodio, J.I.; Snyder, S.H.; Paul, B.D. Regulators of the Transsulfuration Pathway. Br. J. Pharmacol. 2019, 176, 583–593.
- Carballal, S.; Vitvitsky, V.; Kumar, R.; Hanna, D.A.; Libiad, M.; Gupta, A.; Jones, J.W.; Banerjee, R. Hydrogen Sulfide Stimulates Lipid Biogenesis from Glutamine That Is Dependent on the Mitochondrial NAD(P)H Pool. J. Biol. Chem. 2021, 297, 100950.
- Libiad, M.; Vitvitsky, V.; Bostelaar, T.; Bak, D.W.; Lee, H.J.; Sakamoto, N.; Fearon, E.; Lyssiotis, C.A.; Weerapana, E.; Banerjee, R. Hydrogen Sulfide Perturbs Mitochondrial Bioenergetics and Triggers Metabolic Reprogramming in Colon Cells. J. Biol. Chem. 2019, 294, 12077–12090.
- Vitvitsky, V.; Kumar, R.; Libiad, M.; Maebius, A.; Landry, A.P.; Banerjee, R. The Mitochondrial NADH Pool Is Involved in Hydrogen Sulfide Signaling and Stimulation of Aerobic Glycolysis. J. Biol. Chem. 2021, 296, 100736.
- Fu, M.; Zhang, W.; Wu, L.; Yang, G.; Li, H.; Wang, R. Hydrogen Sulfide (H2S) Metabolism in Mitochondria and Its Regulatory Role in Energy Production. Proc. Natl. Acad. Sci. USA 2012, 109, 2943–2948.
- Sen, U.; Pushpakumar, S.B.; Amin, M.A.; Tyagi, S.C. Homocysteine in Renovascular Complications: Hydrogen Sulfide Is a Modulator and Plausible Anaerobic ATP Generator. Nitric Oxide 2014, 41, 27–37.
- Freemerman, A.J.; Johnson, A.R.; Sacks, G.N.; Milner, J.J.; Kirk, E.L.; Troester, M.A.; Macintyre, A.N.; Goraksha-Hicks, P.; Rathmell, J.C.; Makowski, L. Metabolic Reprogramming of Macrophages: Glucose Transporter 1 (GLUT1)-Mediated Glucose Metabolism Drives a Proinflammatory Phenotype. J. Biol. Chem. 2014, 289, 7884–7896.
- Cornwell, A.; Fedotova, S.; Cowan, S.; Badiei, A. Cystathionine γ-Lyase and Hydrogen Sulfide Modulates Glucose Transporter Glut1 Expression via NF-ΚB and PI3k/Akt in Macrophages during Inflammation. PLoS ONE 2022, 17, e0278910.
- Badiei, A.; Gieseg, S.; Davies, S.; Othman, M.I.; Bhatia, M. LPS Up-Regulates Cystathionine γ -Lyase Gene Expression in Primary Human Macrophages via NF-ΚB/ERK Pathway. Inflamm. Allergy Drug Targets 2015, 14, 99–104.
- Gaddam, R.R.; Fraser, R.; Badiei, A.; Chambers, S.; Cogger, V.C.; le Couteur, D.G.; Ishii, I.; Bhatia, M. Cystathionine-Gamma-Lyase Gene Deletion Protects Mice against Inflammation and Liver Sieve Injury Following Polymicrobial Sepsis. PLoS ONE 2016, 11, e0160521.
- Chen, Y.H.; Teng, X.; Hu, Z.J.; Tian, D.Y.; Jin, S.; Wu, Y.M. Hydrogen Sulfide Attenuated Sepsis-Induced Myocardial Dysfunction Through TLR4 Pathway and Endoplasmic Reticulum Stress. Front. Physiol. 2021, 12, 653601.
- Li, J.; Li, M.; Li, L.; Ma, J.; Yao, C.; Yao, S. Hydrogen Sulfide Attenuates Ferroptosis and Stimulates Autophagy by Blocking MTOR Signaling in Sepsis-Induced Acute Lung Injury. Mol. Immunol. 2022, 141, 318–327.
- Li, J.; Ma, J.; Li, M.; Tao, J.; Chen, J.; Yao, C.; Yao, S. GYY4137 Alleviates Sepsis-Induced Acute Lung Injury in Mice by Inhibiting the PDGFRβ/Akt/NF-ΚB/NLRP3 Pathway. Life Sci. 2021, 271, 119192.
- Zhou, T.; Qian, H.; Zheng, N.; Lu, Q.; Han, Y. GYY4137 Ameliorates Sepsis-Induced Cardiomyopathy via NLRP3 Pathway. Biochim. Biophys Acta Mol. Basis. Dis. 2022, 1868.
- Lee, Z.W.; Low, Y.L.; Huang, S.; Wang, T.; Deng, L.W. The Cystathionine γ-Lyase/Hydrogen Sulfide System Maintains Cellular Glutathione Status. Biochem. J. 2014, 460, 425–435.
- Ribas, V.; García-Ruiz, C.; Fernández-Checa, J.C. Glutathione and Mitochondria. Front. Pharmacol. 2014, 5 JUL, 151.
- Lingappan, K. NF-ΚB in Oxidative Stress. Curr. Opin. Toxicol. 2018, 7, 81.
- Das, K.C.; Lewis-Molock, Y.; White, C.W. Activation of NF-Kappa B and Elevation of MnSOD Gene Expression by Thiol Reducing Agents in Lung Adenocarcinoma (A549) Cells. Am. J. Physiol. 1995, 269, L588–L602.
- Rojo, A.I.; Salinas, M.; Martín, D.; Perona, R.; Cuadrado, A. Regulation of Cu/Zn-Superoxide Dismutase Expression via the Phosphatidylinositol 3 Kinase/Akt Pathway and Nuclear Factor-KappaB. J. Neurosci. 2004, 24, 7324–7334.
- Schreiber, J.; Jenner, R.G.; Murray, H.L.; Gerber, G.K.; Gifford, D.K.; Young, R.A. Coordinated Binding of NF-KappaB Family Members in the Response of Human Cells to Lipopolysaccharide. Proc. Natl. Acad. Sci. USA 2006, 103, 5899–5904.
- Pompella, A.; Visvikis, A.; Paolicchi, A.; de Tata, V.; Casini, A.F. The Changing Faces of Glutathione, a Cellular Protagonist. Biochem. Pharmacol. 2003, 66, 1499–1503.
- Kimura, Y.; Goto, Y.I.; Kimura, H. Hydrogen Sulfide Increases Glutathione Production and Suppresses Oxidative Stress in Mitochondria. Antioxid Redox Signal 2010, 12, 1–13.
- Kimura, H. Signaling Molecules: Hydrogen Sulfide and Polysulfide. Antioxid Redox Signal 2015, 22, 362.
- Kimura, Y.; Kimura, H. Hydrogen Sulfide Protects Neurons from Oxidative Stress. FASEB J. 2004, 18, 1165–1167.
- Badiei, A.; Muniraj, N.; Chambers, S.; Bhatia, M. Inhibition of Hydrogen Sulfide Production by Gene Silencing Attenuates Inflammatory Activity by Downregulation of NF-ΚB and MAP Kinase Activity in LPS-Activated RAW 264.7 Cells. Biomed Res. Int. 2014, 2014, 848570.
- Sun, F.; Luo, J.H.; Yue, T.T.; Wang, F.X.; Yang, C.L.; Zhang, S.; Wang, X.Q.; Wang, C.Y. The Role of Hydrogen Sulphide Signalling in Macrophage Activation. Immunology 2021, 162, 3.
- Sun, W.H.; Liu, F.; Chen, Y.; Zhu, Y.C. Hydrogen Sulfide Decreases the Levels of ROS by Inhibiting Mitochondrial Complex IV and Increasing SOD Activities in Cardiomyocytes under Ischemia/Reperfusion. Biochem. Biophys. Res. Commun. 2012, 421, 164–169.
- Zhang, H.; Du, J.; Huang, Y.; Tang, C.; Jin, H. Hydrogen Sulfide Regulates Macrophage Function in Cardiovascular Diseases. Antioxid Redox Signal 2023, 38, 45–56.
- Vitvitsky, V.; Kabil, O.; Banerjee, R. High Turnover Rates for Hydrogen Sulfide Allow for Rapid Regulation of Its Tissue Concentrations. Antioxid Redox Signal 2012, 17, 22–31.
- Goubern, M.; Andriamihaja, M.; Nübel, T.; Blachier, F.; Bouillaud, F. Sulfide, the First Inorganic Substrate for Human Cells. FASEB J. 2007, 21, 1699–1706.
- Lagoutte, E.; Mimoun, S.; Andriamihaja, M.; Chaumontet, C.; Blachier, F.; Bouillaud, F. Oxidation of Hydrogen Sulfide Remains a Priority in Mammalian Cells and Causes Reverse Electron Transfer in Colonocytes. Biochim. Biophys. Acta 2010, 1797, 1500–1511.
- Kumar, R.; Landry, A.P.; Guha, A.; Vitvitsky, V.; Lee, H.J.; Seike, K.; Reddy, P.; Lyssiotis, C.A.; Banerjee, R. A Redox Cycle with Complex II Promotes Sulfide Quinone Oxidoreductase Dependent H2S Oxidation. bioRxiv 2021. bioRxiv:2021.09.08.459449.
- Furne, J.; Saeed, A.; Levitt, M.D. Whole Tissue Hydrogen Sulfide Concentrations Are Orders of Magnitude Lower than Presently Accepted Values. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 295, R1479–R1485.
- Levitt, M.D.; Abdel-Rehim, M.S.; Furne, J. Free and Acid-Labile Hydrogen Sulfide Concentrations in Mouse Tissues: Anomalously High Free Hydrogen Sulfide in Aortic Tissue. Antioxid Redox Signal 2011, 15, 373–378.
- Zhou, T.; Liu, W.; Lai, H.; Liu, Y.; Su, W.; Xu, Z. Hydrogen Sulfide Promotes Osteogenesis by Modulating Macrophage Polarization. Int. Immunopharmacol. 2023, 115, 109564.
- Geng, W.; Liu, X.; Tao, B.; He, Y.; Li, K.; Gao, P.; Feng, Q.; Zhao, P.; Luo, Z.; Cai, K. Nitric Oxide Scavenging and Hydrogen Sulfide Production Synergistically Treat Rheumatoid Arthritis. Adv. Healthc. Mater 2023, 12, 2202380.
- Landry, A.P.; Ballou, D.P.; Banerjee, R. Hydrogen Sulfide Oxidation by Sulfide Quinone Oxidoreductase. Chembiochem 2021, 22, 949–960.
- Lohninger, L.; Tomasova, L.; Praschberger, M.; Hintersteininger, M.; Erker, T.; Gmeiner, B.M.K.; Laggner, H. Hydrogen Sulphide Induces HIF-1α and Nrf2 in THP-1 Macrophages. Biochimie 2015, 112, 187–195.