1. Introduction
Overall,
classically activated (M1
) macrophages are effector cells that are induced by interferon (IFN)γ and lipopolysaccharide (LPS), produce pro-inflammatory cytokines and chemokines, as well as reactive oxygen and nitrogen intermediates, and play a role in host defense against pathogens
[1][2][3][2,3,4]. Conversely,
alternatively activated (M2
) macrophages are induced by cytokines, such as IL-4 and IL-10, and play a role in tissue repair, immune regulation, and homeostasis
[4][5]. Recent studies have revealed that hydrogen sulfide (H
2S) acts as a potent inflammatory mediator in macrophages, modulating various activities, including migration, phagocytosis, and cytokine production, suggesting it as a promising therapeutic target for macrophage-mediated inflammatory conditions
[5][6][6,7].
Gasotransmitters are a class of gaseous signaling molecules, including nitric oxide (NO), carbon monoxide (CO), and
hydrogen sulfide (H
2S
). These gases are produced by specific enzymes in cells and serve important functional roles in regulating physiological processes, such as blood flow, inflammation, and cellular respiration
[7][8]. Gasotransmitters differ from traditional signaling molecules, such as hormones and neurotransmitters, in that they freely diffuse through cell membranes to transduce signaling responses directly and indirectly
[8][9]. NO is a potent signaling molecule produced by iNOS that has bactericidal and cytotoxic properties and regulates pro-inflammatory cytokine production and oxidative stress in macrophages. NO has a dual role in cellular physiology, modulating cellular metabolism and acting as a signaling molecule. NO interacts with metal-containing proteins and the electron transport chain, altering cellular respiration and regulating energy production
[9][10]. CO is produced by the action of heme oxygenases (HO-1-3), and its anti-inflammatory role is suggested in macrophages. HO-1 is an enzyme that catalyzes the degradation of heme into biliverdin, iron, and CO
[10][11]. The mechanisms by which gasotransmitters affect macrophage inflammation are not fully understood, yet their therapeutic potential is increasingly recognized.
H
2S is the latest described gasotransmitter and will be the primary focus of this re
svie
archw. H
2S, initially identified as an environmental toxin, is now recognized to act as an autocrine signaling molecule that was discovered to modulate an increasing number of physiological effects
[5][11][6,12]. H
2S is synthesized in mammals via de-sulfuration reactions catalyzed by the enzymes 3-mercaptosulfurtransferase (3-MST), cystathionine β-synthase (CBS), and cystathionine γ-lyase (CSE), and the latter two are enzymes within the transsulfuration pathway (TSP). In basal conditions, TSP shunts homocysteine away from the methionine pathway and towards the cysteine biosynthesis pathway, ultimately leading to the production of glutathione (GSH), an important antioxidant molecule in cells
[11][12][13][12,13,14].
H
2S plays a role in cellular energy metabolism through its involvement in mitochondrial respiration, the Krebs cycle, and glycolysis
[14][15][16][15,16,17]. H
2S is a respiratory toxin at high concentrations and inhibits ATP generation of the mitochondrial electron transport chain (ETC)
[17][18][18,19]. Cellular metabolism regulates the activity of macrophages
[19][20], and H
2S may be implicated in supporting inflammation-induced metabolic rewiring
[20][21]. Increased H
2S production via CSE supports the pro-inflammatory response of macrophages that contributes to inflammatory diseases in in vivo models of sepsis
[21][22][22,23]. However, recent studies utilizing H
2S donors in similar models of induced sepsis note an opposite and anti-inflammatory role of this molecule
[23][24][25][26][24,25,26,27]. These conflicting results underscore the importance of considering the concentration of H
2S and its source in determining its function.
2. Hydrogen Sulfide and Redox Homeostasis
Cellular components are sensitive to the damaging effects of ROS and require increased protective measures to mitigate ROS-triggered DNA damage, mitochondrial and endoplasmic reticulum (ER) stress, and uncontrolled immune response
[12][27][28][13,62,63]. The effect of oxidative stress on NF-κB activation depends on the cell type and context. In response to oxidative stress, NF-κB may induce the expression of several antioxidants
[29][64]. The levels of superoxide dismutase (SOD)
[30][31][65,66] and GPx
[32][67], among others, are increased via NF-κB to protect cellular components from self-inflicted damage and contribute to a regulated immune response.
As the primary mechanism of antioxidant defense against ROS and electrophiles, GSH serves a crucial role in maintaining cellular redox homeostasis
[28][63]. GSH is synthesized
de novo through two ATP-dependent reactions catalyzed by γ-glutamylcysteine synthase and glutathione synthetase. The redox-active thiol (-SH) of cysteine in GSH is oxidized to GSSG when it reduces target molecules and requires NADPH to restore its reduced form
[33][68]. The CSE enzyme plays a crucial role in maintaining the levels of GSH by producing cysteine, the limiting substrate for GSH synthesis
[27][62]. H
2S also increases GSH levels by enhancing cysteine transporter activity
[34][35][69,70] and enhances γ-glutamylcysteine synthase activity, which increases γ-glutamylcysteine levels
[36][71]. NF-κB significantly upregulates CSE during inflammatory response in macrophages
[37][48], and CSE indirectly contributes to antioxidant systems by producing cysteine and H
2S
[34][38][39][69,72,73].
H
2S levels are regulated to support inflammatory responses and can transduce signaling pathways that lead to metabolic rewiring. H
2S can also interact with various signaling molecules, such as ROS and NO, which modulate macrophage inflammation potentially reducing oxidative stress in specific disease context including cardiovascular diseases
[40][74]. Additionally, H
2S can directly affect cellular metabolism by inhibiting mitochondrial respiration and promoting glycolysis, which can lead to a rewiring of metabolic pathways
[15][41][16,75]. H
2S activity upon mitochondrial bioenergetics is bi-phasic. The electron transport chain couples redox reactions, creating an electrochemical gradient that leads to the creation of ATP. H
2S stimulates mitochondrial respiration at lower concentrations but inhibits it at higher concentrations
[15][42][43][16,76,77]. Elevated H
2S levels are potent ETC inhibitors via its binding activities upon the metallic center in complex IV
[15][16]. However, at physiologic levels, H
2S elevates oxygen consumption rates and electron flow through ETC via
sulfide quinone oxidoreductase (SQR)SQR [15][16]. Recent evidence challenges the conventional view that H
2S only exerts toxic effects in cells; in these conditions where the electron transport chain is obstructed, high levels of H
2S can continue to undergo oxidation by SQR, leading to a redox cycle with fumarate as the terminal electron acceptor at complex II
[44][78].
Alternatively, in the event of inhibition of the ETC by excessive H
2S, increased glycolytic flux is triggered by increased cytosolic NADH levels
[16][17]. In response, lactate production is elevated through increased lactate dehydrogenase activity, which serves as a counterbalance. This creates a redox-neutral cycle, allowing the cell to continue producing ATP through glycolysis despite ETC inhibition. The cycle relies on the functioning of the electrogenic glutamate–aspartate transporter, which plays a vital role in the malate–aspartate shuttle
[16][17]. This interaction demonstrates how H
2S may signal to influence cellular energy metabolism, and place increased glucose substrate demands on cells exposed to H
2S. When mitochondria are under reductive pressure due to an elevated NADH pool, the normally oxidizing Krebs cycle may become reprogrammed. Indeed, in H
2S-treated colonocytes, the citrate level was increased due to the reversal of the Krebs cycle via the stimulation of glutamine-dependent reductive carboxylation
[15][16]. Overall, the multilevel regulation of TSP enzymes, coupled with mitochondrial SQR demonstrates a remarkable sulfur sensitive redox system in place that regulates numerous cellular systems including cellular metabolism. Since these systems are highly regulated and induced in macrophages during inflammation, future research is required to elucidate these systems in these cells, which may be targeted to reduce disease severity.
3. The Therapeutic Potential of Hydrogen Sulfide and Polysulfides
H
2S is known for its ability to scavenge reactive oxygen and nitrosyl species, although endogenous levels of H
2S (10–30 nM) are too low for direct free-radical scavenging
[45][46][79,80], exogenous H
2S donors, however, may achieve elevated levels. GYY4137, a slow-releasing H
2S donor, has demonstrated anti-inflammatory effects and protective properties against various conditions, such as septic peritonitis, acute lung injury, and myocardial injury. In M0/M1 macrophages, GYY4137 treatment increased the expression of CD-206 and IL-10 while reducing iNOS and TNF-α expression in M1 macrophages. When combined with IL-4, GYY4137 treatment enhanced M0 macrophage viability and mineralized particle formation in cocultures with MC3T3-E1 osteoblasts
[47][81]. In sepsis studies, GYY4137 notably alleviated septic peritonitis and protected against cecal ligation and puncture (CLP)-induced acute lung injury by decreasing neutrophil infiltration, ameliorating sepsis-induced lung histopathological alterations, and lessening lung injury severity. This protective effect was associated with reduced levels of PDGFRβ, NF-κB, ASC, NLRP3, caspase-1, and Akt proteins in septic mouse lung tissues
[25][26].
GYY4137 is also reported to mitigate ferroptosis and inhibit autophagy activation in macrophage. This compound reduced macrophage infiltration in septic heart tissue and protected against myocardial injury via the NLRP3 inflammasome pathway by decreasing inflammatory response and myocardial ROS production
[26][47][27,81]. Furthermore, GYY4137 alleviated ferroptosis in septicemia-induced ALI by upregulating GPx and SLC7A11 expression in lung tissue post-CLP. Upon LPS exposure, the expression of mTOR, P62, and Beclin1 increased, while the LC3II/LC3I ratio decreased; however, GYY4137 treatment proved protective by blocking mTOR signaling and inhibiting autophagy activation
[24][25].
Recent studies have explored novel H
2S donor polymers or polysulfide donors, observing similar protective effects. One such novel polymer, which scavenges NO and donates H
2S, has been shown to decrease ROS levels and pro-inflammatory cytokine production via NF-κB signaling, promoting macrophage M2 polarization and H
2S release. In a rat model of rheumatoid arthritis, this system significantly reduced synovial inflammation, osteoporosis, and clinical symptoms
[48][82]. Collectively, these findings suggest that H
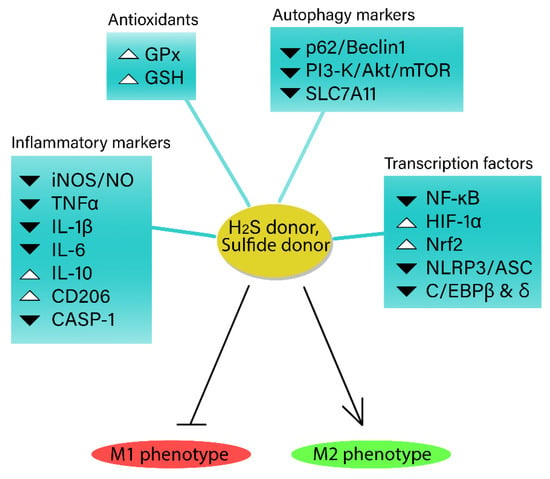
2S and its donors hold promise as therapeutic agents for various inflammatory conditions diverting macrophage to the anti-inflammatory phenotype (
Figure 1).
Figure 1. The role of exogenous hydrogen sulfide (H2S) in macrophage inflammatory phenotype. H2S helps regulate inflammatory responses, while also interacting with pro-inflammatory markers, transcription factors, and proteins to modulate macrophage inflammation. H2S donors have demonstrated potential therapeutic benefits in various inflammatory conditions, characterized by inducing anti-inflammatory M2 phenotype in macrophages, highlighting their promise as treatment options.
4. The Complex Role of Hydrogen Sulfide in Modulating Cellular Energy Metabolism and Inflammatory Responses in Macrophages
H
2S plays a key role in regulating cellular energy metabolism and redox homeostasis, which is important for regulating inflammation in macrophages, but its precise role on immune cell inflammatory response is not yet fully understood. Due to the susceptibility of respiration to H
2S toxicity at high levels, the di-flavin enzyme, SQR, which is localized on the inner mitochondrial membrane, acts as a respiratory shield. H
2S attacks the disulfide bond in SQR, generating a persulfide charge transfer complex. This outer sulfane sulfur is moved to a thiophilic acceptor, such as GSH, safeguarding ETC function from H
2S
[41][44][49][75,78,103]. H
2S orchestrates regulatory pathways in cellular bioenergetics, modulating mitochondrial energy production and promoting glycolysis. Previously, it was reported that treatment with the H
2S-donor molecule, GYY4137, induced the expression of Glut1 by stabilizing HIF-1α under normal oxygen conditions in THP-1 macrophages
[50][104]. This same study also reported that prolonged exposure (24 h) of unstimulated macrophage to high levels of exogenous H
2S decreased NF-κB and increased Nrf2 activity.
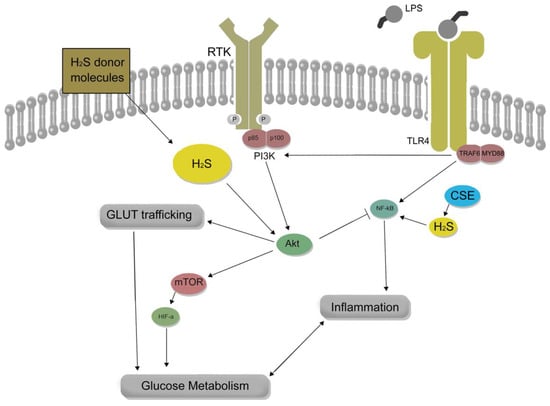
ResearchWe
rs previously reported the role of endogenous H
2S to support inflammatory Glut1 expression in macrophages via its regulatory role upon NF-κB and Akt activity
[20][21] (
Figure 2). Thus, H
2S plays a vital role in macrophage function by regulating cellular energy metabolism and redox homeostasis, influencing inflammation, and impacting various regulatory pathways that modulate mitochondrial energy production and promote glycolysis.
The regulatory function of H2S in mitochondrial respiration has become a focal point of discussion, owing to its potential to modulate cellular bioenergetics. Despite the uncertainty surrounding the effects of H2S on the inflammatory responses of immune cells, recent investigations have illuminated the association between H2S and cellular energy metabolism. H2S demonstrates a biphasic impact on cellular energy metabolism and may be implicated in the reconfiguration of inflammatory metabolism. This elaborate interplay underscores the capacity of H2S to influence cellular energy metabolism and impose heightened glucose substrate demands on cells exposed to H2S. Nevertheless, this relationship remains poorly understood, necessitating additional research to unravel the specific mechanisms underpinning this intricate association and the consequences of these effects in various pathological conditions. The interaction between exogenous and endogenous H2S and glucose metabolism presents a captivating area of exploration. H2S seemingly fosters a favorable redox environment for glycolysis, alluding to the possibility of further crosstalk. Intriguingly, H2S has also been found to induce alterations in transcription factors and proteins, bolstering the expression of inflammatory Glut1 in macrophages. The present resviearchw emphasizes the importance of continued research to disentangle the complex interrelations between H2S and cellular energy metabolism, ultimately unveiling novel insights into the broader implications of this signaling molecule.
Figure 2. The role of hydrogen sulfide (H
2S) in macrophage inflammation and glucose metabolism is complex and context-dependent. H
2S donor molecules can enhance lipopolysaccharide (LPS)-induced Akt activity, which may counteract pro-inflammatory NF-κB activity. Conversely, cystathionine-γ-lyase (CSE)-derived H
2S supports NF-κB pro-inflammatory activity, leading to the expression of glucose transporter 1 (Glut1) and metabolic reprogramming in macrophages during inflammation. The impact of H
2S on macrophage inflammation, therefore, depends on factors such as the source, concentration, and context of H
2S exposure, which can result in either pro- or anti-inflammatory outcomes.