Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Beatrix Zheng and Version 1 by György Trencsényi.
As malignancies still represent one of the major health concerns worldwide, early tumor identification is among the priorities of today’s science. Given the strong association between cyclooxygenase-2 (COX-2)/prostaglandin E2 (PGE2), PGE2 receptors (EPs), and carcinogenesis, target-specific molecules directed towards the components of the COX2/PGE2/EP axis seem to be promising imaging probes in the diagnostics of PGE2pos. neoplasms and in the design of anti-cancer drugs.
- Bismuth-205/206 (205/206Bi)
- cyclodextrins (CDs)
- Gallium-68 (68Ga)
- 2-hydroxypropyl-β-cyclodextrin (HPBCD)
1. Cyclooxigenase-2 (COX2)/Prostaglandin E2 (PGE2)/PGE2-Receptor (EP) Axis
Given the strong association between cyclooxygenase-2 (COX-2)/prostaglandin E2 (PGE2), PGE2 receptors (EPs), and carcinogenesis, an immense number of research studies has been spawned to intensively investigate the comprehensive molecular mechanisms behind [1].
Membrane-attached, inducible COX-2 is the key rate-limiting enzyme in the conversion of arachidonic acid into prostaglandins [1]. Literature data indicate that COX-2 overexpression seems critical in cancer initiation, tumor progression, and tumor maintenance [2]. Further, prior studies reported COX-2 upregulation in different cancer types, such as glioblastoma, colon, breast, prostate, and urinary bladder cancer [3,4,5,6,7][3][4][5][6][7]. Hence, COX2 specific inhibitors (COXibs) or non-steroidal anti-inflammatory drugs (NSAIDs) that block the activity of COX2 could contribute to the mitigation of cancer-linked mortality [8]. For example, Harris et al. published that celecoxib and rofecoxib effectively reduced the risk of development of colon cancer in a study conducted with the enrolment of 326 patients with colon cancer of invasive type and 652 disease naïve control subjects [9]. Added to that, the feasibility of COX2 inhibitor NSAIDs in tumor growth prevention and the lengthening of survival were further confirmed at preclinical level [10].
COX2 together with the microsomal prostaglandin E2 synthase-1 (mPGES-1) are the two major contributors to PGE2 synthesis through the prostanoid biosynthetic pathway [1]. In addition, PGE2 concentration is also influenced by the activity of 15-hydroxyprostaglandin dehydrogenase (15-PGDH)—the main catalysator of the oxidisation of PGE2 to inert 15-keto-PGE2 [11,12][11][12]. Beyond the synthesis processes and the degradation of PGE2, both the intra- and the intercellular PGE2 levels are mediated by drug resistance-associated protein (MRP4) and prostaglandin uptake transporter (PGT) [13,14][13][14].
Identically to COX2, there is a notable interplay between the excess production of PGE2 and tumorigenesis [15]. PGE2 acting via G-protein coupled rhodopsin-type prostanoid receptors E 1–4 (EP1, EP2, EP3, EP4) triggers various tumor-associated intracellular signaling pathways, out of which the most remarkable ones are the stimulation of the epidermal growth factor-receptor (EGFR), the phosphorylation of protein kinase C (PKC)-mediated extracellular signal-regulated kinase (ERK), and the activation of G-protein dependent beta-catenin [1,2,12,16,17][1][2][12][16][17]. The PGE2 regulated activation of EP receptor induces tumor-related angiogenesis along with the enhancement of tumor cell invasion and the halting of natural immune responses [12]. Further, PGE2-generated tumor proliferation as well as apoptosis inhibition lead to tumor volume expansion [12]. Former translational studies registered moderate tumor propagation in EP2-knockout mice in comparison with the wild-type ones [18]. Recent research indicating a link between 15-PGDH down-regulation and the appearance of certain malignancies including lung and transitional bladder cancers may project the role of this degrading enzyme in carcinogenesis [19,20][19][20]. Moreover, the metastatic spread of triple negative breast cancer was promoted by MRP4 [21]. In addition, Tong and colleagues claimed that PGE2 may serve as a biomarker in different malignancies such as pancreatic, breast, oral, or renal cancer [1].
Target-specific molecules directed towards the components of the COX2/PGE2/EP axis seem to be promising imaging probes in the early diagnostics of PGE2pos. neoplastic alterations as well as in the design of anti-cancer drug candidates. Nuclear medical in vivo preclinical model systems applying target-selective radiolabelled compounds may aid to further understand the molecular pathways of COX2/PGE2 that would attribute to the identification of well-suited diagnostic and anti-tumor biomarkers.
In the present revisearch thew we researchers provide a detailed overview about PGE2- specific radiolabelled diagnostic probes and their potential therapeutic applications in PGE2 overexpressing malignancies.
2. PGE2 and Cyclodextrins (CDs)
Cyclodextrins (CDs) are arising as powerful diagnostic agents in the field of nuclear medicine. Built up by D-(+)-glucopyranose molecules, these truncated cone-shaped oligosaccharides have a hydrophobic inner cage and a hydrophilic outer surface [22,23,24,25][22][23][24][25]. Given their advantageous physicochemical characteristic features, CDs are highly valuable for forming inclusion complexes with a myriad of molecules including PGE2. The complex forming capability of β-CDs with PGE2 was already established by Hirayama et al. in 1984 [26]. Exploiting the beneficial biochemical characteristics of β-CDs, Hirayama and co-workers managed to obtain the stabilization of PGE2 in aqueous solution with the application of the following two modified β-CD derivatives: heptakis (2, 6-di-O-methyl)-β-CD (DM-β-CD) and heptakis (2, 3, 6-tri-O-methyl)-β-CD (TM-β-CD). Compared to TM-β-CD, DM-β-CD appeared to be more effective in the maintenance of the stability of PGE2 projecting its feasibility in the proposal of pharmaceutical dosage formulas [26].
Besides the above-mentioned CD compounds, the complexation of another β-CD derivative—randomly methylated β-cyclodextrin (RAMEB)—with PGE2 was exhaustively evaluated as well. In a recent in vitro study focusing on the interaction between RAMEB and various proinflammatory molecules such as PGE2, substance P (SP), bradykinin (BRK), and calcitonin gene-related peptide (CGRP), Sauer and co-workers observed that PGE2 displays more significant binding potential to RAMEB in comparison with the other investigated agents [27]. To indicate the best fitting β-CD guest molecule, the Molecular Operating Environment docking algorithm-based molecular docking of SP, CGRP, BRK, and PGE2 was carried out. While RAMEB-affine PGE2 (ΔGbind= −12.57 kcal/moL) was characterised by high CD binding capability, BRK was moderately connected to RAMEB (−10.54 kcal/moL). Neither of the remaining assessed molecules showed affinity towards RAMEB. Earlier studies also focused on the evaluation of the complexation between CDs and other PGE subtypes, such as prostaglandin E1 (PGE1). Inoue et al. managed to obtain the enhanced stability of PGE1 derivative Limaprost/α-CD complex (Limaprost-alfadex) in humid circumstances by adding β-CD to the existing agent [28]. Later, they investigated the way of complexation between the PGE1 compound and the CD molecules, applying 1H- and 13C-Nuclear Magnetic Resonance (NMR) spectroscopy [29]. Referring to their results, a kind of ternary complex formation could be accountable for the stabilization of the drug. Overall, their findings also strengthened the strong association between CDs and PGE molecules.
Based on the above remarked former discoveries, PGE2-affine CDs appear to be valuable diagnostic vectors and drug carrier vehicles in PGE2-associated pathological processes, including carcinogenesis. Moreover, radiolabelled CDs complexed with PGE2 may establish the basis of target-specific diagnostics of PGE2 expressing malignancies. Applying radiometals with therapeutic radiation for labelling purposes, the theranostic potential of PGE2-directed CDs could also be tested (Figure 1). Prior to the definitive integration of PGE2-affine CD-based imaging probes into routine clinical use, the need for the verification of their in vivo diagnostic applicability is warranted. Preclinical small animal model systems ensure appropriate circumstances for the assessment of the stability, the pharmacokinetics, the biodistribution, and the tumor-homing effectiveness of radiolabelled CD compounds.
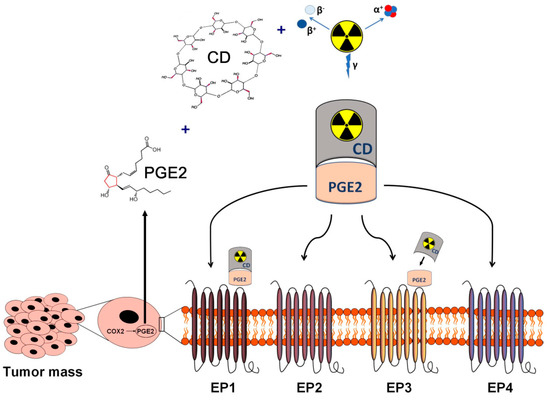
Figure 1. Schematic overview of the in vivo diagnostics and radioisotope therapy of PGE2-expressing and PGE receptor-positive tumors using radiolabelled CDs. Applying radioisotopes with diagnostic (γ) and therapeutic radiation (α or β), radiolabelled CDs complexed with tumor cell derived PGE2 may establish the basis of target-specific diagnostics and personalized radionuclide therapy of PGE2-expressing malignancies. CD: cyclodextrin; PGE2: prostaglandin E2.
Positron emission tomography (PET) is the mainstay diagnostic device in the detection of primary tumors and pertinent metastases. PET purveys data on in vivo physiological and pathological processes with the application of various targeted radiopharmaceuticals containing positron emitting radioisotopes [30]. The non-invasive characteristics and high sensitivity of PET imaging make it well-suited for the preclinical evaluation of tumor-selective PGE2-specific molecules [31,32][31][32].
In 2019, Hajdu and his colleagues launched a novel Gallium-68 (68Ga)-labelled, p-NCS-benzyl-NODA-GA (NODAGA)-conjugated β-CD compound—denoted as 68Ga-NODAGA-(2-hydroxypropyl)-β-cyclodextrin ([68Ga]Ga-NODAGA-HPBCD)—to verify its physicochemical adequacy for potential future in vivo PET diagnostics [31]. This research could be hailed as a landmark study in the proposal of further radiolabelled CD-based derivatives for diagnostic purposes. In their study they elaborated the radiochemical synthesis process of this newly constructed PET probe. The exceptional radiochemical purity (RCP; 96–99%) (defined by radio-HPLC), the hydrophilic physicochemical property (LogP = −3.07 ± 0.11), and the stability in mouse serum confirmed the feasibility of [68Ga]Ga-NODAGA-HPBCD in upcoming in vivo investigations. Applying healthy 12-week-old male BALB/c mice in vivo PET/CT examinations indicated a renal way of excretion along with low tracer uptake values measured 90 min postinjection in the liver (SUVmean: 0.12 ± 0.03), the intestines (SUVmean: 0.11 ± 0.02), the heart (SUVmean: 0.13 ± 0.02), the lung (SUVmean: 0.17 ± 0.02), and the brain (SUVmean: 0.09 ± 0.02). The ex vivo biodistribution data were in line with the in vivo figures. Since the established synthesis process led to the production of an intravenously (iv.) applicable radiotracer with adequate organ distribution and outstanding chemical features, it seemed to be adaptable for the manufacturing of additional radio-conjugated CD molecules. Hence, CD-based, PGE2-specific PET radiopharmaceuticals (Figure 2) could be established for cancer imaging on the basis of the synthesis model of Hajdu et al. Table 1. displays the in vivo preclinical studies with PGE2-affine radiolabelled imaging probes.
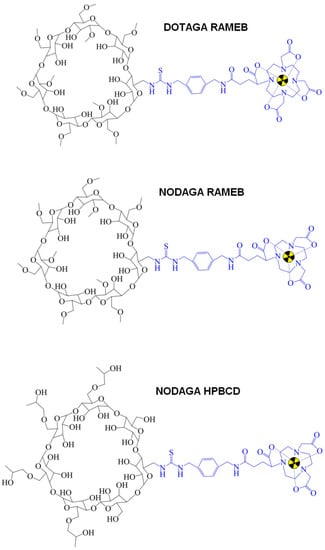
Figure 2. Chemical structure of CD-based PET radiopharmaceuticals. CD: cyclodextrin; PET: positron emission tomography.
Table 1. Overview of preclinical studies with radiolabelled prostaglandin E2 (PGE2)-targeting cyclodextrins (CDs).
| Investigated Object | Investigated Phenomenon | Radiopharmaceutical | Imaging Technique | Reference |
|---|---|---|---|---|
| healthy BALB/c male mice | diagnostic feasibility, radiochemical synthesis process, proof-of-concept study | [68Ga]Ga-NODAGA-HPBCD | in vivo PET/CT imaging, ex vivo radioactivity determination by gamma counter | Hajdu et al., 2019 [31] |
| BxPC-3 and PancTu-1 tumor-bearing CB17 SCID mice | in vivo and ex vivo organ distribution, PGE2 tumor-homing ability | [68Ga]Ga-NODAGA-RAMEB | in vivo static and dynamic PET imaging, ex vivo gamma counter measurements | Trencsényi et al., 2020 [33] |
| CB17 SCID male mice bearing BxPC-3 tumor | ex vivo biodistribution, PGE2-targeting capability, pharmacokinetic profile, biosynthesis | [68Ga]Ga-DOTAGA-RAMEB [205/206Bi]Bi-DOTAGA-RAMEB |
in vivo static and dynamic PET acquisition, ex vivo gamma counting | Csige et al., 2022 [34] |
| HT1080, -A20, -PancTu-1, -BxPC-3, -B16-F10 tumor-bearing CB17 SCID male mice and Ne/De, He/De tumor-bearing Fischer-344 female rats | in vivo and ex vivo biodistribution pattern, tumor-targeting competence | [68Ga]Ga-NODAGA-RAMEB, [68Ga]Ga-NODAGA-HPBCD |
in vivo static PET imaging, ex vivo gamma counting | Szabó et al., 2023 [35] |
A20: mouse B cell lymphoma; 205/206Bi: Bismuth-205/206; B16-F10: mouse melanoma; BxPC-3: human pancreas adenocarcinoma; CD: cyclodextrin; DOTAGA: 1,4,7,10-tetrakis(carboxymethyl)-1,4,7,10-tetraazacyclododecane glutaric acid; PancTu-1: human pancreas adenocarcinoma; 68Ga: Gallium-68; He/De: rat hepatocellular carcinoma; HPBCD: 2-hydroxypropyl-β-cyclodextrin; HT1080: human fibrosarcoma; Ne/De: rat mesoblastic nephroma; NODAGA: 1,4,7-triazacyclononane-1-glutaric acid-4,7-diacetic acid; PET/CT: positron emission tomography/computed tomography; PGE2: prostaglandin E2; RAMEB: randomly methylated β-cyclodextrin; SCID: severe combined immunodeficient.
3. PGE2pos. Preclinical Tumor Models
BxPC-3 pancreatic adenocarcinoma cell lines are firmly associated with elevated PGE2 synthesis and EP2 receptor upregulation [36]. Investigating the following pancreatic cell lines in vitro: MiaPaCa-2, BxPC-3, PANC-1, and Capan-1, Takahashi and co-workers observed the expression of EP and various growth factor receptors by reverse transcriptase-polymerase chain reaction (RT-PCR). EIA-based (Enzyme immunoassay) determination of PGE2 levels and the presence of COX1 and COX2 mRNA were also evaluated. Applying EIA, ten-fold higher PGE2 concentrations were observed in BxPC-3 culture media relative to the other investigated specimens. Moderate EP2 mRNA presence and enhanced EP4 mRNA expression were identified in BxPC-3 cells. Further, comparing all investigated cells lines, the most remarkable COX- and EP4-reliant autocrine loop was manifested in the BxPC-3 culture.
The discrete presence of COX2 enzyme and related inconsiderable PGE2 synthesis of Panc-Tu-1 pancreatic ductal adenocarcinoma (PDAC) cells was reported by Gonnermann et al. [37]. Four PDAC cells lines including PancTu-1, Pt45P1, Panc89, and Colo357 were analysed in their study. As part of the comparison analyses of the PDAC cell cultures, disproportions between the appearance of COX2 and COX2-generated PGE2 synthesis were determined. In accordance with the PGE2 levels, in PancTu-1 and in Panc89 cells both flow-cytometry- and Western Blot-based measurements revealed moderate COX2 presence in comparison with receptor upregulated Colo357 cell lines. Less than 0.5 ng/mL PGE2 was produced by PancTu-1 and Panc89 cells, while Colo357-associated synthesis exceeded 6 ng/mL. In a like manner, Szabó et al. also confirmed the low EP2 receptorial expression of PancTu-1 cells during the immunohistochemical verification of the presence of EP2 receptor in several cell lines [35].
To outhe researchers' best knowledge, no exact literature data is available regarding the EP2 receptor status of HT1080 cells; however, according to the results of some earlier studies, the existence of these receptors and pertinent PGE2 production may be concluded. Anti-Prostaglandin E Receptor EP2 antibody immunohistochemistry with 3,3-diaminobenzidine (DAB) chromogens was employed to detect the appearance of EP2 receptor on HT1080 cells [35]. Histological staining revealed low amount of membrane EP2 receptors on the cell surface of HT1080 cell lines. Pifithrin-α (PFT-α)-induced COX2 level enhancement experienced in HT1080 cells with p53 wild-type suggests the presence of both PGE2 and its receptors in fibrosarcoma cell lines [38]. P-53 transactivation inhibitor PTF-α triggers COX2 overexpression via mitogen-activated protein kinase (MAPK) kinase (MEK)/extracellular signal-regulated kinase (ERK)-based molecular mechanisms. In a bid to investigate PFT-α-generated COX2 overexpression, wild-type p53 HT1080 fibrosarcoma cells were administered with 40 µmol/L PFT-α. COX2 presence was authenticated applying Western blot analyses. Compared to the control, HT1080 cells exhibited 5.3-fold higher COX2 presence after PFT-α application.
Pi et al. established an anti-programmed cell death protein 1 (anti-PD-1) drug-resistant B16-F10 (B16-F10-R) preclinical melanoma Pdcd1 transgenic mouse model system to investigate the expression of programmed death-ligand 1 (PD-L1) and lymphocyte invasion into the tumor niche [39]. Additionally, associations between COX2 and anti-PD-1 pembrolizumab resistance were surveyed as well. At an average tumor bulk of 50 mm3, beyond the iv. administration of 10 mg/BW (kg) pembrolizumab (anti-PD-1 antibody) or its control type twice a week, aspirin (10 mg/BW), SC560 (selective COX1 inhibitor, 5 mg/BW), celecoxib (selective COX2 inhibitor, 5 mg/BW), and E7046 (EP4 inhibitor, 10 mg/BW) were intraperitoneally injected three times weekly to the following tumor-bearing experimental small animal groups: B16-F10, B16-F10-NR (anti-PD-1 non-resistant), B16-F10-R (anti-PD-1 resistant), and B16-F10-R-knockout ptgs2 (B16-F10-knockout prostaglandin endoperoxide synthases/COX2). Prostaglandin endoperoxide synthases 1/prostaglandin endoperoxide synthases2 (Ptgs1/Ptgs2, COX1/COX2) transcript levels and PGE2 values were determined applying Real-Time Quantitative Polymerase Chain Reaction (RT-qPCR) and Enzyme-Linked Immunosorbent Assay (ELISA). Amongst others, anti-COX2 antibody was utilized for the accomplishment of Western Blot analyses, while T and natural killer (NK) cells were identified by flow cytometry-based staining. B10-F10-R malignancies were featured with enhanced PD-L1 expression-associated reduced CD3+, CD4+, and CD8+ T cell and NK cell invasion, as well as the overexpression of the ptgs2 gene. Albeit these phenomena seemed to be reversible with the co-administration of non-selective COX1/COX2 inhibitor and anti-PD-1 antibody. Upregulated COX2 levels with associated PGE2 synthesis characterised the B16-F10-R tumor-bearing mice, reflecting the major contribution of COX2 to anti-cancer drug sensitivity. Further, anti-tumor drug insensitivity was abolished utilizing ptgs2 knockout or E7046 selective EP4 inhibitor combined with anti-PD-1 treatment. Given the COX2 axis inhibition-originated heightened immune cell invasion (T and NK cells) as well as therapeutic drug-sensitivity wthe researchers presume the role of EP2 receptors and PGE2 in the biological processes of B16-F10 cell lines.
B-cell-associated immunoglobulin production is regulated by PGE2 in immunological-allergic processes [40]. Of note, 9 times higher serum IgE concentrations and much elevated monocyte-driven PGE2 synthesis characterised the patients suffering from progressive Hodgkin’s disease (HD). Previous literature data confirm the PGE2-related EP receptor positivity of B lymphocytes [41,42,43][41][42][43]. Further, in vivo investigation of the receptor profile of mouse B lymphocytes revealed the presence of type 2 and type 4 EP receptors on the surface of the cell membrane (EP2, EP4) [44]. Taking the B-cell origin of HD into consideration, these findings may imply the strong EP receptor expression of B cells and different B cell lines.
More recently, chemically-induced, rat-originated syngeneic Ne/De (rat mesoblastic nephroma) and He/De (rat hepatocellular carcinoma) tumors and their metastases also proved to be PGE2pos. during preclinical PET imaging studies [35].
References
- Tong, D.; Liu, Q.; Wang, L.A.; Xie, Q.; Pang, J.; Huang, Y.; Wang, L.; Liu, G.; Zhang, D.; Lan, W.; et al. The roles of the COX2/PGE2/EP axis in therapeutic resistance. Cancer Metastasis Rev. 2018, 37, 355–368.
- Wu, W.K.; Sung, J.J.; Lee, C.W.; Yu, J.; Cho, C.H. Cyclooxygenase-2 in tumorigenesis of gastrointestinal cancers: An update on the molecular mechanisms. Cancer. Lett. 2010, 295, 7–16.
- Banu, N.; Buda, A.; Chell, S.; Elder, D.; Moorghen, M.; Paraskeva, C.; Qualtrough, D.; Pignatelli, M. Inhibition of COX-2 with NS-398 decreases colon cancer cell motility through blocking epidermal growth factor receptor transactivation: Possibilities for combination therapy. Cell Prolif. 2007, 40, 768–779.
- Cook, P.J.; Thomas, R.; Kingsley, P.J.; Shimizu, F.; Montrose, D.C.; Marnett, L.J.; Tabar, V.S.; Dannenberg, A.J.; Benezra, R. Cox-2-derived PGE2 induces Id1-dependent radiation resistance and self-renewal in experimental glioblastoma. Neuro. Oncol. 2016, 18, 1379–1389.
- Kim, H.S.; Moon, H.G.; Han, W.; Yom, C.K.; Kim, W.H.; Kim, J.H.; Noh, D.Y. COX2 overexpression is a prognostic marker for stage III breast cancer. Breast. Cancer Res. Treat. 2012, 132, 51–59.
- Liu, Q.; Yuan, W.; Tong, D.; Liu, G.; Lan, W.; Zhang, D.; Xiao, H.; Zhang, Y.; Huang, Z.; Yang, J.; et al. Metformin represses bladder cancer progression by inhibiting stem cell repopulation via COX2/PGE2/STAT3 axis. Oncotarget 2016, 7, 28235–28246.
- Tong, D.; Liu, Q.; Liu, G.; Xu, J.; Lan, W.; Jiang, Y.; Xiao, H.; Zhang, D.; Jiang, J. Metformin inhibits castration-induced EMT in prostate cancer by repressing COX2/PGE2/STAT3 axis. Cancer Lett. 2017, 389, 23–32.
- Chan, A.T.; Ogino, S.; Fuchs, C.S. Aspirin use and survival after diagnosis of colorectal cancer. JAMA 2009, 302, 649–658.
- Harris, R.E.; Beebe-Donk, J.; Alshafie, G.A. Similar reductions in the risk of human colon cancer by selective and nonselective cyclooxygenase-2 (COX-2) inhibitors. BMC. Cancer 2008, 8, 237.
- Yao, M.; Zhou, W.; Sangha, S.; Albert, A.; Chang, A.J.; Liu, T.C.; Wolfe, M.M. Effects of nonselective cyclooxygenase inhibition with low-dose ibuprofen on tumor growth, angiogenesis, metastasis, and survival in a mouse model of colorectal cancer. Clin. Cancer Res. 2005, 11, 1618–1628.
- Tai, H.H.; Ensor, C.M.; Tong, M.; Zhou, H.; Yan, F. Prostaglandin catabolizing enzymes. Prostaglandins Other Lipid Mediat. 2002, 68–69, 483–493.
- Wang, D.; Dubois, R.N. Prostaglandins and cancer. Gut 2006, 55, 115–122.
- Reid, G.; Wielinga, P.; Zelcer, N.; van der Heijden, I.; Kuil, A.; de Haas, M.; Wijnholds, J.; Borst, P. The human multidrug resistance protein MRP4 functions as a prostaglandin efflux transporter and is inhibited by nonsteroidal antiinflammatory drugs. Proc. Natl. Acad. Sci. USA 2003, 100, 9244–9249.
- Kochel, T.J.; Fulton, A.M. Multiple drug resistance-associated protein 4 (MRP4), prostaglandin transporter (PGT), and 15-hydroxyprostaglandin dehydrogenase (15-PGDH) as determinants of PGE2 levels in cancer. Prostaglandins Other Lipid Mediat. 2015, 116–117, 99–103.
- Brown, J.R.; DuBois, R.N. COX-2: A molecular target for colorectal cancer prevention. J. Clin. Oncol. 2005, 23, 2840–2855.
- Madrigal-Martínez, A.; Fernández-Martínez, A.B.; Lucio Cazaña, F.J. Intracrine prostaglandin E2 pro-tumoral actions in prostate epithelial cells originate from non-canonical pathways. J. Cell. Physiol. 2018, 233, 3590–3602.
- Pan, J.; Yang, Q.; Shao, J.; Zhang, L.; Ma, J.; Wang, Y.; Jiang, B.H.; Leng, J.; Bai, X. Cyclooxygenase-2 induced β1-integrin expression in NSCLC and promoted cell invasion via the EP1/MAPK/E2F-1/FoxC2 signal pathway. Sci. Rep. 2016, 6, 33823.
- Ma, X.; Aoki, T.; Tsuruyama, T.; Narumiya, S. Definition of prostaglandin E2-EP2 signals in the colon tumor microenvironment that amplify inflammation and tumor growth. Cancer Res. 2015, 75, 2822–2832.
- Ding, Y.; Tong, M.; Liu, S.; Moscow, J.A.; Tai, H.H. NAD+-linked 15-hydroxyprostaglandin dehydrogenase (15-PGDH) behaves as a tumor suppressor in lung cancer. Carcinogenesis 2005, 26, 65–72.
- Gee, J.R.; Montoya, R.G.; Khaled, H.M.; Sabichi, A.L.; Grossman, H.B. Cytokeratin 20, AN43, PGDH, and COX-2 expression in transitional and squamous cell carcinoma of the bladder. Urol. Oncol. 2003, 21, 266–270.
- Kochel, T.J.; Reader, J.C.; Ma, X.; Kundu, N.; Fulton, A.M. Multiple drug resistance-associated protein (MRP4) exports prostaglandin E2 (PGE2) and contributes to metastasis in basal/triple negative breast cancer. Oncotarget 2017, 8, 6540–6554.
- Del Valle, E.M.M. Cyclodextrins and their uses: A review. Process Biochem. 2004, 39, 1033–1046.
- Loftsson, T.; Jarho, P.; Másson, M.; Järvinen, T. Cyclodextrins in drug delivery. Expert. Opin. Drug. Deliv. 2005, 2, 335–351.
- Szejtli, J. Cyclodextrins and Their INCLUSION Complexes; Academic Publisher: Budapest, Hungary, 1982; p. 395.
- Szejtli, J. Introduction and General Overview of Cyclodextrin Chemistry. Chem. Rev. 1998, 98, 1743–1754.
- Hirayama, F.; Kurihara, M.; Uekama, K. Improving the aqueous stability of prostaglandin E2 and prostaglandin A2 by inclusion complexation with methylated-beta-cyclodextrins. Chem. Pharm. Bull. 1984, 32, 4237–4240.
- Sauer, R.S.; Rittner, H.L.; Roewer, N.; Sohajda, T.; Shityakov, S.; Brack, A.; Broscheit, J.A. A Novel Approach for the Control of Inflammatory Pain: Prostaglandin E2 Complexation by Randomly Methylated β-Cyclodextrins. Anesth. Analg. 2017, 124, 675–685.
- Inoue, Y.; Sekiya, N.; Katayama, K.; Narutaki, S.; Yamamoto, M.; Iohara, D.; Hirayama, F.; Uekama, K. Stabilizing effect of β-cyclodextrin on Limaprost, a PGE1 derivative, in Limaprost alfadex tablets (Opalmon) in highly humid conditions. Chem. Pharm. Bull. 2014, 62, 786–792.
- Inoue, Y.; Sekiya, N.; Yamamoto, M.; Iohara, D.; Hirayama, F.; Uekama, K. Formation of the ternary inclusion complex of limaprost with α- and β-cyclodextrins in aqueous solution. Chem. Pharm. Bull. 2015, 63, 318–325.
- Phelps, M.E. Positron emission tomography provides molecular imaging of biological processes. Proc. Natl. Acad. Sci. USA 2000, 97, 9226–9233.
- Hajdu, I.; Angyal, J.; Szikra, D.; Kertész, I.; Malanga, M.; Fenyvesi, É.; Szente, L.; Vecsernyés, M.; Bácskay, I.; Váradi, J.; et al. Radiochemical synthesis and preclinical evaluation of 68Ga-labeled NODAGA-hydroxypropyl-beta-cyclodextrin (68Ga-NODAGA-HPBCD). Eur. J. Pharm. Sci. 2019, 128, 202–208.
- Phelps, M.E. PET: The merging of biology and imaging into molecular imaging. J. Nucl. Med. 2000, 41, 661–681.
- Trencsényi, G.; Kis, A.; Szabó, J.P.; Ráti, Á.; Csige, K.; Fenyvesi, É.; Szente, L.; Malanga, M.; Méhes, G.; Emri, M.; et al. In vivo preclinical evaluation of the new 68Ga-labeled beta-cyclodextrin in prostaglandin E2 (PGE2) positive tumor model using positron emission tomography. Int. J. Pharm. 2020, 576, 118954.
- Csige, K.; Szabó, J.P.; Kálmán-Szabó, I.; Dénes, N.S.; Szikra, D.; Képes, Z.; Opposits, G.; Méhes, G.; Kertész, I.; Fenyvesi, F.; et al. In vivo investigation of Gallium-68 and Bismuth-205/206 labeled beta cyclodextrin for targeted alpha therapy of prostaglandin E2 receptor-expressing tumors in mice. Int. J. Pharm. 2022, 625, 122132.
- Szabó, J.P.; Csige, K.; Kálmán-Szabó, I.; Arató, V.; Opposits, G.; Jószai, I.; Kertész, I.; Képes, Z.; Méhes, G.; Fenyvesi, F.; et al. In vivo assessment of tumor targeting potential of 68Ga-labelled randomly methylated beta-cyclodextrin (RAMEB) and 2-hydroxypropyl-β-cyclodextrin (HPβCD) using positron emission tomography. Int. J. Pharm. 2023, 630, 122462.
- Takahashi, T.; Uehara, H.; Ogawa, H.; Umemoto, H.; Bando, Y.; Izumi, K. Inhibition of EP2/EP4 signaling abrogates IGF-1R-mediated cancer cell growth: Involvement of protein kinase C-θ activation. Oncotarget 2015, 6, 4829–4844.
- Gonnermann, D.; Oberg, H.H.; Kellner, C.; Peipp, M.; Sebens, S.; Kabelitz, D.; Wesch, D. Resistance of cyclooxygenase-2 expressing pancreatic ductal adenocarcinoma cells against γδ T cell cytotoxicity. Oncoimmunology 2015, 4, e988460.
- Kim, S.; Han, J.; Lee, S.K.; Hur, S.M.; Koo, M.; Cho, D.H.; Bae, S.Y.; Choi, M.Y.; Shin, I.; Yang, J.H.; et al. Pifithrin-α, an inhibitor of p53 transactivation, up-regulates COX-2 expression through an MAPK-dependent pathway. Pharmacology 2010, 86, 313–319.
- Pi, C.; Jing, P.; Li, B.; Feng, Y.; Xu, L.; Xie, K.; Huang, T.; Xu, X.; Gu, H.; Fang, J. Reversing PD-1 Resistance in B16F10 Cells and Recovering Tumour Immunity Using a COX2 Inhibitor. Cancers 2022, 14, 4134.
- Fedyk, E.R.; Brown, D.M.; Phipps, R.P. PGE2 regulation of B lymphocytes and T helper 1 and T helper 2 cells: Induction of inflammatory versus allergic responses. Adv. Exp. Med. Biol. 1997, 407, 237–242.
- Brown, D.M.; Phipps, R.P. Characterization and regulation of prostaglandin E2 receptors on normal and malignant murine B lymphocytes. Cell. Immunol. 1995, 161, 79–87.
- Burchiel, S.W.; Hanson, K.; Warner, N.L. Clonal heterogeneity of cyclic AMP responsiveness: A comparison of malignant murine lymphoid cell lines. Int. J. Immunopharmacol. 1984, 6, 35–42.
- Roper, R.L.; Phipps, R.P. Prostaglandin E2 and cAMP inhibit B lymphocyte activation and simultaneously promote IgE and IgG1 synthesis. J. Immunol. 1992, 149, 2984–2991.
- Fedyk, E.R.; Ripper, J.M.; Brown, D.M.; Phipps, R.P. A molecular analysis of PGE receptor (EP) expression on normal and transformed B lymphocytes: Coexpression of EP1, EP2, EP3beta and EP4. Mol. Immunol. 1996, 5, 33–45.
More