Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Rita Xu and Version 1 by Jehad Abubaker.
Obstructive sleep apnoea (OSA) is a prevalent underdiagnosed disorder whose incidence increases with age and weight. Uniquely characterised by frequent breathing interruptions during sleep—known as intermittent hypoxia (IH)—OSA disrupts the circadian rhythm. Patients with OSA have repeated episodes of hypoxia and reoxygenation, leading to systemic consequences. OSA consequences range from apparent symptoms like excessive daytime sleepiness, neurocognitive deterioration and decreased quality of life to pathological complications characterised by elevated biomarkers linked to endocrine-metabolic and cardiovascular changes. OSA is a well-recognized risk factor for cardiovascular and cerebrovascular diseases.
- obstructive sleep apnoea
- inflammation
- cardiovascular disease
1. Introduction
Impacting approximately 25% of men and 13% of women, and with a prevalence of 56% worldwide [1,2,3][1][2][3], obstructive sleep apnoea (OSA) is characterised by the partial or complete collapse of the upper airway, preventing normal ventilation during sleep. As a result, individuals with OSA often feel fatigued and suffer from daytime sleepiness, lack of focus, reduced vigilance, memory impairment and overall reduced quality of life (QoL). Although male sex, advanced age and obesity are known to increase the likelihood of developing OSA, other variables such as race/ethnicity, family history and craniofacial dysmorphisms also play a role in developing OSA [4]. Additional signs and symptoms of OSA include snoring, choking and gasping during sleep, nocturia, insomnia and morning headaches [5,6][5][6]. Untreated OSA results in frequent arousal and desaturation during sleep. As a result, OSA leads to fragmented sleep and recurrent episodes of intermittent hypoxia (IH) and reoxygenation. Long-lasting hypoxemia and the resulting hypoxia can have severe health consequences, increasing the risk of developing cardiovascular diseases, including systemic hypertension, atherosclerosis, coronary artery disease, stroke and metabolic conditions such as insulin resistance and type 2 diabetes (T2D) [7,8][7][8]. In addition, OSA is associated with cognitive impairment, dementia, depression and motor vehicle accidents due to reduced daytime alertness [9,10][9][10]. Obesity is the primary risk factor that can be altered to reduce the likelihood of developing OSA; even modest weight control was successful in reducing the occurrence of sleep-disordered breathing, according to a population-based cohort analysis of 690 subjects. The study found that for every 10% increase in body weight, the apnoea-hypopnoea index (AHI) increased by nearly 32% [11].
There is an association between OSA and expanding waistlines and necks, thus OSA risk factors include a neck circumference greater than 17 inches in males and 16 inches in women. After controlling for BMI, neck circumference still appears to be a predictor of OSA, and it may even provide a stronger correlation with specific measures of disease severity in patients with OSA. Smoking, a family history of OSA and nocturnal nasal congestion are additional, less well-established risk factors. Among the various treatment methods for OSA, continuous positive airway pressure (CPAP) is the first-line treatment for patients with OSA and excessive daytime sleepiness due to its high efficacy rates [12,13][12][13]. Furthermore, CPAP is one of the best management options for relieving symptoms of classic OSA and improving the QoL of patients [14]. Additionally, the use of CPAP was associated with improvements in cognitive ability [15[15][16],16], insulin resistance [17,18][17][18] and reductions in blood pressure in patients with OSA and resistant hypertension [19,20][19][20]. On the other hand, a recent meta-analysis showed that the use of CPAP in patients with OSA was neither associated with reduced cardiovascular outcomes and death nor improved glycemic control in patients with type 2 diabetes [21,22,23,24][21][22][23][24].
2. OSA and Inflammation
The continuous stress caused by recurrent snoring and repetitive closure and re-opening of the upper airway in patients with OSA can lead to mucosa inflammation [26,27][25][26]. Low-grade inflammation causes remodelling of the upper airway, with increased deposition of connective tissue in the mucosa and muscles. Consequently, introducing changes to the morphology of palatopharyngeal muscle, inflammation and denervation contribute to upper airway obstruction during sleep in patients with OSA [28,29][27][28]. Therefore, systemic and localised inflammation induced by IH and the repeated cycles of hypoxia and reoxygenation (Figure 1) features several events: induction of oxidative stress, reactive oxygen species (ROS) production [30][29], activation of a critical proinflammatory transcription factor (nuclear factor κB (NF-kB)) [31][30], elevated expression of proinflammatory cytokines and chemokines [32[31][32],33], recruitment and infiltration of the proinflammatory M1 macrophages [30,34][29][33] in different tissues such as vessels, heart, adipose tissue and liver. Eventually, this contributes to vascular remodelling, metabolic dysfunction [35][34] and atherosclerosis [36][35].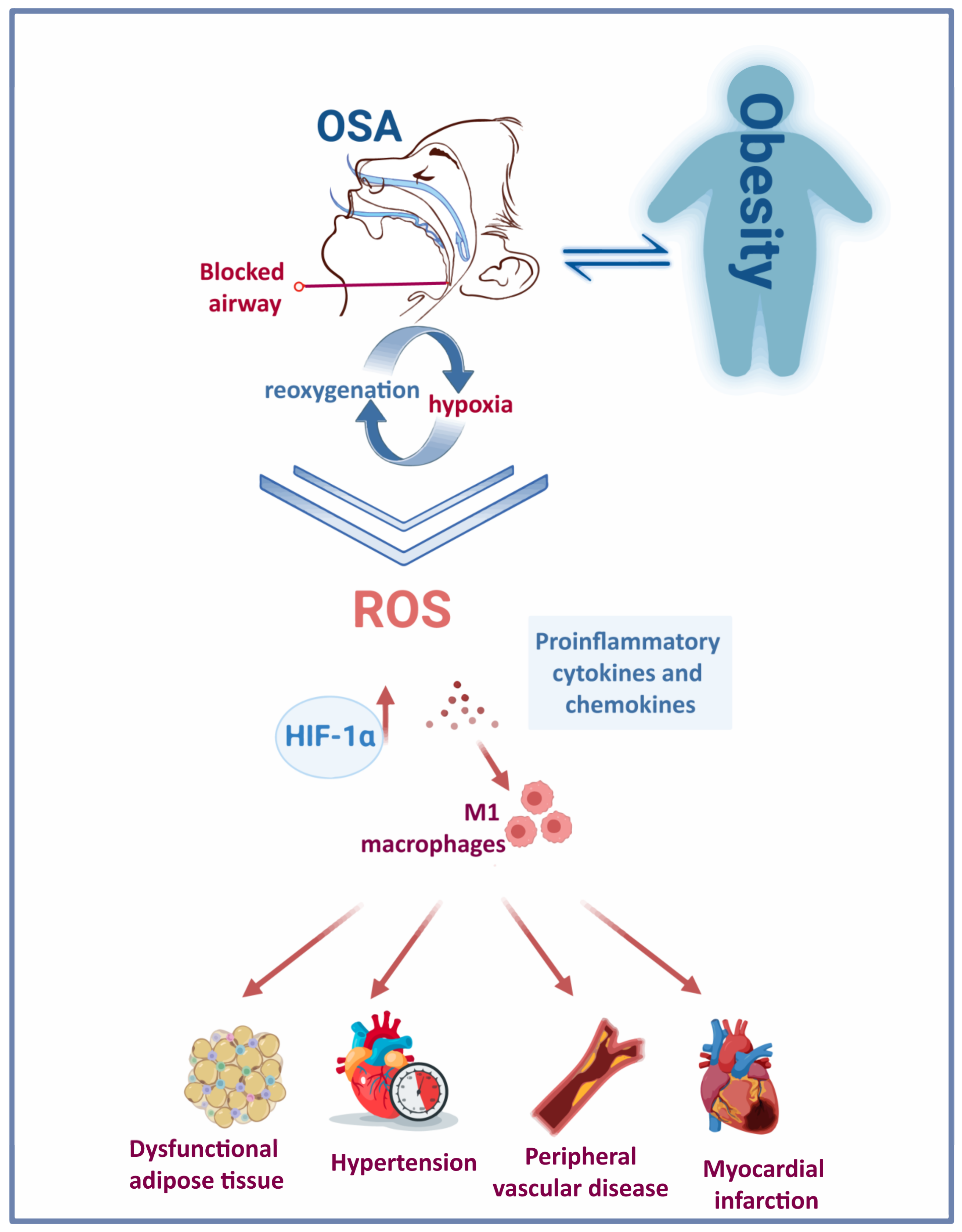
Figure 1. Obesity and OSA: a bidirectional interaction. The repetitive cycles of hypoxia-reoxygenation due to frequent blockage of the upper airway in people with OSA triggers the activation of various pathological mechanisms and induces a rise in HIF-1α. The chronic presence of this condition has deleterious consequences on the cardiovascular system.
3. OSA and Obesity
OSA is intimately associated with obesity, with OSA being diagnosed in more than 30% of people with obesity and 50–98% of people with morbid obesity [73][51]; thus, a higher BMI score increases the likelihood of developing OSA [74][52]. Both conditions are associated with severe cardio-metabolic complications. The rising prevalence of OSA in the past 20 years concurred with increasing prevalence of obesity worldwide [75][53]. Obesity is an established major risk factor for OSA [74,76][52][54]. Three extensive cohort studies have shown that weight gain is associated with an increased risk of developing moderate to severe OSA and correlated with increased AHI. In contrast, weight loss coalesced with reduced AHI and OSA severity [11,77,78][11][55][56]. Several reports proposed obesity as a factor contributing to the pathogenesis of OSA by increasing fat deposition in the parapharyngeal fat pads and tongue, thus narrowing the upper airway [79,80][57][58]. Furthermore, obesity can cause chest and abdominal wall compression, reducing tracheal tension and contributing to a more collapsible upper airway [76,81][54][59]. Obesity was associated with the severity of blood oxygen desaturation during apnoea and hypopnoea, thus likely aggravating OSA symptoms in these patients [82][60]. On the other hand, the relationship between OSA and obesity may be reciprocal, as OSA may also contribute to weight gain. Many patients have reported gaining weight on the onset of OSA diagnosis [83,84][61][62]. Several aspects of OSA may contribute to the development or aggravation of obesity: the fragmented sleep pattern associated with OSA affects dietary habits and sleep duration, leading to hormonal changes that affect satiety and energy expenditure [85][63]. Increased daytime sleepiness and fatigue also contribute to reduced physical activity, leading to weight gain and increased risk of obesity comorbidities [73][51]. CPAP therapy may also impact body weight in patients with OSA. Meta-analyses of randomised controlled trials showed that CPAP therapy led to a small but significant increase in body mass index [35][34], particularly in high-usage patients [86][64]. However, these analyses involved studies of relatively short duration (median of 3 months). Contrastingly, a post hoc analysis of the Sleep Apnea Cardiovascular Endpoints (SAVE) cohort showed that long-term use of CPAP (>3 years) did not have a significant impact on body weight in patients with comorbid OSA and cardiovascular disease [87][65]. Notably, both OSA and obesity trigger the secretion of proinflammatory factors. An increasing body of evidence supports the common pathways by which obesity and OSA-associated IH lead to inflammation and dysfunction of adipose tissue (Figure 1). However, given the similar mechanisms that trigger low-grade inflammation in obesity and OSA, discerning between OSA-specific and obesity-triggered inflammatory biomarkers remains challenging [88][66]. In addition, proinflammatory cells are targeted at dysfunctional adipose tissue, producing proinflammatory factors such as TNF-α, IL-6, IL-1β, monocyte chemotactic protein (MCP)-1, resistin and leptin [67]. Levels of the proinflammatory cytokines IL-17 and IL-23 were significantly elevated in paediatric OSA cases [69][68]. In a study involving children with obesity and OSA, IL-17 was significantly associated with OSA, while IL-23 levels correlated with body fat and liver enzymes. Therefore, studies proposed these proinflammatory cytokines as potential biomarkers of paediatric OSA [70][69]. Additionally, leptin levels significantly increased in people with sleep disorder breathing. The rise in leptin levels correlated directly with BMI Z-score, AHI [70,71,72][69][70][71] and fasting insulin [70][69]. The relationship between OSA and cytokines was corroborated by an observational study showing the diurnal variation of IL-6, IL-8 and TNF-α with the expression of symptoms in patients with mild OSA [68][72]. Repeated exposure to hypoxia alters gene transcription and posttranslational protein modification, influencing metabolic and cardiovascular processes. OSA and obesity both trigger hypoxia-inducible factor 1 (HIF-1) in adipocytes, ultimately stimulating the expression of downstream angiogenic proteins to increase oxygen and nutrient delivery to adipocytes. Primarily responsible for maintaining stable oxygen metabolism, HIF-1 is an attractive target that is selectively induced by chronic IH and promoted by oxidative stress. The heterodimeric complex comprises two different proteins: HIFα and HIFβ. Both belong to a family of transcription factors constitutively expressed in cells [89][73]. HIF-1α is oxygen-sensitive and couples with von Hippel-Lindau protein during normoxia to trigger its degradation by proteasomes [90][74]. The degradation of HIF-1α is inhibited under hypoxic conditions [91][75]. It is estimated that HIF-1 activates over 100 distinct genes [92][76]. Consequently, HIF-1 is a critical transcription factor that regulates numerous processes, including the metabolism and cardiovascular system [92,93][76][77]. However, many signalling pathways in which it participates remain poorly understood. Among numerous others, it stimulates genes associated with angiogenesis or glucose uptake by cells, i.e., glucose transporter 1 and glucose transporter 4 [92,94][76][78].4. OSA and Cardiovascular Disease
OSA is a recognised independent risk factor for CVD and is associated with metabolic abnormalities that are closely linked to cardiovascular diseases. In accordance with this, OSA is associated with hypertension [104,105,106][79][80][81], which puts patients with OSA at increased risk of developing cardiovascular complications such as coronary artery disease and stroke [107][82]. Hypertension affects 36.5–53.6% of patients with OSA, depending on severity of the condition [108,109][83][84]. Mechanisms driving increased blood pressure in patients with OSA are well described. Obstruction of the upper airways leads to hypoxemia, hypoxia, hypercapnia and changes in intrathoracic pressure, which, combined with frequent arousal from sleep, induces sympathetic system activation, increasing both heart rate and blood pressure. These hemodynamic changes contribute to tachycardia and hypertension, which can develop into left ventricular hypertrophy and heart failure [107][82]. Chronic OSA is linked to oxidative stress due to the elevated synthesis of ROS brought on by the frequent episodes of IH and reoxygenation, which contribute to the development of oxidative stress. Several cross-sectional studies have examined different oxidative stress markers in patients with OSA. For example, high-sensitivity CRP, metalloproteinase 9 and copper correlate with higher AHI and lower haemoglobin oxygen saturation [110,111][85][86]. Similar results were reported with the protein disulfide reductase thioredoxin [112,113][87][88]. In addition, Malondialdehyde, a common biomarker of lipid peroxidation, has also been associated with OSA, correlating with the duration of nocturnal oxygen desaturation below 85% [114,115,116][89][90][91]. Furthermore, patients with OSA presented with higher levels of oxidised LDL compared with healthy controls [117,118][92][93]. Additionally, the presence of OSA concurred with lower ferric-reducing antioxidant power (FRAP) compared with controls, which negatively correlated with AHI [119,120][94][95]. On the other hand, several oxidative stress biomarkers did not correlate with OSA: 8-isoprostane, thiobarbituric acid-reactive substances, erythrocyte catalase activity, copper-zinc superoxide dismutase (SOD) and total antioxidant capacity [121,122][96][97]. While the benefit of CPAP therapy in reducing oxidative stress biomarkers in patients with OSA is debatable [56[98][99],123], antioxidant therapy is effective in reducing oxidative stress and is a potential alternative to CPAP in patients with OSA [124,125,126][100][101][102]. Several studies have investigated polyphenols as a treatment for OSA. Grebe et al. reported improved endothelial health through measurements of flow-mediated dilation (FMD) of the brachial artery. This study reported lower FMD measurements in a group of patients with OSA after receiving vitamin C intravenously compared with controls [127][103]. Additionally, vitamins C and E have been studied for their potential to mitigate oxidative stress in laboratory rodents and other animal models. Higher levels of malondialdehyde (MDA) and advanced oxidation protein products (AOPP), both markers of oxidative stress, were found in subjects who experienced intermittent hypoxia caused by tracheal obstruction. Although the levels of MDA were not affected by the administration of antioxidants, AOPP levels were reduced considerably [128][104]. Antioxidant properties are found in several medications commonly used to treat diseases other than OSA. N-acetylcysteine (NAC) is a well-known mucolytic drug that is used to treat acetaminophen overdose, is required for glutathione synthesis and has antioxidant properties. In a study involving patients with OSA, oral administration of NAC for 30 days caused a significant reduction in lipid peroxidation products and significantly increased glutathione levels [124][100]. Currently, an ongoing clinical trial (NCT05009901) is examining the effect of using a powerful oral antioxidant (alpha lipoic acid, ALA) to treat patients with OSA by improving their cardiovascular health and reducing systemic inflammation and markers of oxidative stress. Another possible mechanism for the development of hypertension and consequent cardiovascular disease in patients with OSA is the activation of the renin-angiotensin-aldosterone system (RAAS). Meta-analyses of 13 studies assessing the role of OSA on RAAS components have reported higher plasma levels of angiotensin II in patients with OSA compared with controls and that patients with hypertension and OSA had higher aldosterone plasma levels compared with the controls [129][105]. IH and reoxygenation may also contribute to the development of cardiovascular disease by promoting inflammation, which is strongly associated with endothelial dysfunction, atherosclerosis and coronary artery disease [130][106]. Numerous animal and cell-based studies have pointed to a strong link between IH and vascular and systemic inflammation development. In mice, IH-induced atherosclerotic changes occurred with increased expression of proinflammatory cytokines, chemokines and adhesion molecules, increased migration of inflammatory cells and expansion of the macrophage population in the arterial wall [31,36,131][30][35][107]. In addition, the aorta of mice exposed to IH had higher expression of the proinflammatory transcription factor NF-kB, although the levels of NF-kB returned to normal after recovering from normoxia [32,132][31][108]. In humans, monocytes of patients with severe OSA showed elevated NLRP3 activity compared with monocytes from control subjects and this elevation showed a direct correlation with the AHI score and other hypoxemic indices [57][109]. This report showed that higher NLRP3 activity triggered inflammatory cytokines, i.e., IL-1β and IL-18, via caspase-1 and increased Gasdermin D, which consequently allowed for tissue factor to be released. As a result, plasma concentrations of tissue factor were higher in patients with OSA and systemic inflammatory comorbidities compared with controls [57][109]. In a prospective cohort study, the activation of NF-κB coincided with endothelial dysfunction and higher levels of its downstream targets were detected in endothelial cells of patients with OSA [133][110]. In particular, the effects on endothelial function were reversed after four weeks of CPAP therapy [134][111].References
- Chen, Y.C.; Hsu, P.Y.; Hsiao, C.C.; Lin, M.C. Epigenetics: A Potential Mechanism Involved in the Pathogenesis of Various Adverse Consequences of Obstructive Sleep Apnea. Int. J. Mol. Sci. 2019, 20, 2937.
- Andayeshgar, B.; Janatolmakan, M.; Soroush, A.; Azizi, S.M.; Khatony, A. The prevalence of obstructive sleep apnea in patients with type 2 diabetes: A systematic review and meta-analysis. Sleep Sci. Pract. 2022, 6, 6.
- Benjafield, A.V.; Ayas, N.T.; Eastwood, P.R.; Heinzer, R.; Ip, M.S.M.; Morrell, M.J.; Nunez, C.M.; Patel, S.R.; Penzel, T.; Pépin, J.L.; et al. Estimation of the global prevalence and burden of obstructive sleep apnoea: A literature-based analysis. Lancet Respir. Med. 2019, 7, 687–698.
- Chen, X.; Wang, R.; Zee, P.; Lutsey, P.L.; Javaheri, S.; Alcántara, C.; Jackson, C.L.; Williams, M.A.; Redline, S. Racial/Ethnic Differences in Sleep Disturbances: The Multi-Ethnic Study of Atherosclerosis (MESA). Sleep 2015, 38, 877–888.
- Ouayoun, M.C.; Chabolle, F.; De Vito, A.; Heiser, C.; Paramasivan, V.K.; Rabelo, F.A.W.; Rotenberg, B.; Suurna, M.V. International consensus (ICON) on the ENT role in diagnosis of obstructive sleep apnea syndrome. Eur. Ann. Otorhinolaryngol. Head Neck Dis. 2018, 135, S3–S6.
- Veasey, S.C.; Rosen, I.M. Obstructive Sleep Apnea in Adults. N. Engl. J. Med. 2019, 380, 1442–1449.
- Garvey, J.F.; Pengo, M.F.; Drakatos, P.; Kent, B.D. Epidemiological aspects of obstructive sleep apnea. J. Thorac. Dis. 2015, 7, 920–929.
- Sarkar, P.; Mukherjee, S.; Chai-Coetzer, C.L.; McEvoy, R.D. The epidemiology of obstructive sleep apnoea and cardiovascular disease. J. Thorac. Dis. 2018, 10, S4189–S4200.
- Osman, A.M.; Carter, S.G.; Carberry, J.C.; Eckert, D.J. Obstructive sleep apnea: Current perspectives. Nat. Sci. Sleep 2018, 10, 21–34.
- Epstein, L.J.; Kristo, D.; Strollo, P.J., Jr.; Friedman, N.; Malhotra, A.; Patil, S.P.; Ramar, K.; Rogers, R.; Schwab, R.J.; Weaver, E.M.; et al. Clinical guideline for the evaluation, management and long-term care of obstructive sleep apnea in adults. J. Clin. Sleep Med. JCSM Off. Publ. Am. Acad. Sleep Med. 2009, 5, 263–276.
- Peppard, P.E.; Young, T.; Palta, M.; Dempsey, J.; Skatrud, J. Longitudinal study of moderate weight change and sleep-disordered breathing. JAMA 2000, 284, 3015–3021.
- Cao, M.T.; Sternbach, J.M.; Guilleminault, C. Continuous positive airway pressure therapy in obstuctive sleep apnea: Benefits and alternatives. Expert Rev. Respir. Med. 2017, 11, 259–272.
- Jat, K.R.; Mathew, J.L. Continuous positive airway pressure (CPAP) for acute bronchiolitis in children. Cochrane Database Syst. Rev. 2019, 1, Cd010473.
- Lo Bue, A.; Salvaggio, A.; Iacono Isidoro, S.; Romano, S.; Insalaco, G. OSA and CPAP therapy: Effect of gender, somnolence, and treatment adherence on health-related quality of life. Sleep Breath. Schlaf Atm. 2019, 24, 533–540.
- Davies, C.R.; Harrington, J.J. Impact of Obstructive Sleep Apnea on Neurocognitive Function and Impact of Continuous Positive Air Pressure. Sleep Med. Clin. 2016, 11, 287–298.
- Olaithe, M.; Bucks, R.S. Executive dysfunction in OSA before and after treatment: A meta-analysis. Sleep 2013, 36, 1297–1305.
- Iftikhar, I.H.; Khan, M.F.; Das, A.; Magalang, U.J. Meta-analysis: Continuous positive airway pressure improves insulin resistance in patients with sleep apnea without diabetes. Ann. Am. Thorac. Soc. 2013, 10, 115–120.
- Feng, Y.; Zhang, Z.; Dong, Z.Z. Effects of continuous positive airway pressure therapy on glycaemic control, insulin sensitivity and body mass index in patients with obstructive sleep apnoea and type 2 diabetes: A systematic review and meta-analysis. NPJ Prim. Care Respir. Med. 2015, 25, 15005.
- Liu, L.; Cao, Q.; Guo, Z.; Dai, Q. Continuous Positive Airway Pressure in Patients With Obstructive Sleep Apnea and Resistant Hypertension: A Meta-Analysis of Randomized Controlled Trials. J. Clin. Hypertens. 2016, 18, 153–158.
- Lei, Q.; Lv, Y.; Li, K.; Ma, L.; Du, G.; Xiang, Y.; Li, X. Effects of continuous positive airway pressure on blood pressure in patients with resistant hypertension and obstructive sleep apnea: A systematic review and meta-analysis of six randomized controlled trials. J. Bras. Pneumol. Publicacao Soc. Bras. Pneumol. Tisilogia 2017, 43, 373–379.
- Labarca, G.; Reyes, T.; Jorquera, J.; Dreyse, J.; Drake, L. CPAP in patients with obstructive sleep apnea and type 2 diabetes mellitus: Systematic review and meta-analysis. Clin. Respir. J. 2018, 12, 2361–2368.
- Da Silva Paulitsch, F.; Zhang, L. Continuous positive airway pressure for adults with obstructive sleep apnea and cardiovascular disease: A meta-analysis of randomized trials. Sleep Med. 2019, 54, 28–34.
- Yu, J.; Zhou, Z.; McEvoy, R.D.; Anderson, C.S.; Rodgers, A.; Perkovic, V.; Neal, B. Association of Positive Airway Pressure with Cardiovascular Events and Death in Adults With Sleep Apnea: A Systematic Review and Meta-analysis. JAMA 2017, 318, 156–166.
- Azarbarzin, A.; Zinchuk, A.; Wellman, A.; Labarca, G.; Vena, D.; Gell, L.; Messineo, L.; White, D.P.; Gottlieb, D.J.; Redline, S.; et al. Cardiovascular Benefit of Continuous Positive Airway Pressure in Adults with Coronary Artery Disease and Obstructive Sleep Apnea without Excessive Sleepiness. Am. J. Respir. Crit. Care Med. 2022, 206, 767–774.
- Salerno, F.G.; Carpagnano, E.; Guido, P.; Bonsignore, M.R.; Roberti, A.; Aliani, M.; Vignola, A.M.; Spanevello, A. Airway inflammation in patients affected by obstructive sleep apnea syndrome. Respir. Med. 2004, 98, 25–28.
- Inancli, H.M.; Enoz, M. Obstructive sleep apnea syndrome and upper airway inflammation. Recent Pat. Inflamm. Allergy Drug Discov. 2010, 4, 54–57.
- Boyd, J.H.; Petrof, B.J.; Hamid, Q.; Fraser, R.; Kimoff, R.J. Upper airway muscle inflammation and denervation changes in obstructive sleep apnea. Am. J. Respir. Crit. Care Med. 2004, 170, 541–546.
- Lindman, R.; Stål, P.S. Abnormal palatopharyngeal muscle morphology in sleep-disordered breathing. J. Neurol. Sci. 2002, 195, 11–23.
- Dewan, N.A.; Nieto, F.J.; Somers, V.K. Intermittent hypoxemia and OSA: Implications for comorbidities. Chest 2015, 147, 266–274.
- Gras, E.; Belaidi, E.; Briançon-Marjollet, A.; Pépin, J.-L.; Arnaud, C.; Godin-Ribuot, D. Endothelin-1 mediates intermittent hypoxia-induced inflammatory vascular remodeling through HIF-1 activation. J. Appl. Physiol. 2016, 120, 437–443.
- Arnaud, C.; Beguin, P.C.; Lantuejoul, S.; Pepin, J.L.; Guillermet, C.; Pelli, G.; Burger, F.; Buatois, V.; Ribuot, C.; Baguet, J.P.; et al. The inflammatory preatherosclerotic remodeling induced by intermittent hypoxia is attenuated by RANTES/CCL5 inhibition. Am. J. Respir. Crit. Care Med. 2011, 184, 724–731.
- Maniaci, A.; Iannella, G.; Cocuzza, S.; Vicini, C.; Magliulo, G.; Ferlito, S.; Cammaroto, G.; Meccariello, G.; De Vito, A.; Nicolai, A. Oxidative stress and inflammation biomarker expression in obstructive sleep apnea patients. J. Clin. Med. 2021, 10, 277.
- Murphy, A.M.; Thomas, A.; Crinion, S.J.; Kent, B.D.; Tambuwala, M.M.; Fabre, A.; Pepin, J.L.; Roche, H.M.; Arnaud, C.; Ryan, S. Intermittent hypoxia in obstructive sleep apnoea mediates insulin resistance through adipose tissue inflammation. Eur. Respir. J. 2017, 49, 1601731.
- Drager, L.F.; Brunoni, A.R.; Jenner, R.; Lorenzi-Filho, G.; Benseñor, I.M.; Lotufo, P.A. Effects of CPAP on body weight in patients with obstructive sleep apnoea: A meta-analysis of randomised trials. Thorax 2015, 70, 258–264.
- Arnaud, C.; Poulain, L.; Lévy, P.; Dematteis, M. Inflammation contributes to the atherogenic role of intermittent hypoxia in apolipoprotein-E knock out mice. Atherosclerosis 2011, 219, 425–431.
- Paulsen, F.P.; Steven, P.; Tsokos, M.; Jungmann, K.; Müller, A.; Verse, T.; Pirsig, W. Upper airway epithelial structural changes in obstructive sleep-disordered breathing. Am. J. Respir. Crit. Care Med. 2002, 166, 501–509.
- Sekosan, M.; Zakkar, M.; Wenig, B.L.; Olopade, C.O.; Rubinstein, I. Inflammation in the uvula mucosa of patients with obstructive sleep apnea. Laryngoscope 1996, 106, 1018–1020.
- Vicente, E.; Marin, J.M.; Carrizo, S.J.; Osuna, C.S.; González, R.; Marin-Oto, M.; Forner, M.; Vicente, P.; Cubero, P.; Gil, A.V.; et al. Upper airway and systemic inflammation in obstructive sleep apnoea. Eur. Respir. J. 2016, 48, 1108–1117.
- Chua, A.P.; Aboussouan, L.S.; Minai, O.A.; Paschke, K.; Laskowski, D.; Dweik, R.A. Long-term continuous positive airway pressure therapy normalizes high exhaled nitric oxide levels in obstructive sleep apnea. J. Clin. Sleep Med. JCSM Off. Publ. Am. Acad. Sleep Med. 2013, 9, 529–535.
- Fortuna, A.M.; Miralda, R.; Calaf, N.; González, M.; Casan, P.; Mayos, M. Airway and alveolar nitric oxide measurements in obstructive sleep apnea syndrome. Respir. Med. 2011, 105, 630–636.
- Culla, B.; Guida, G.; Brussino, L.; Tribolo, A.; Cicolin, A.; Sciascia, S.; Badiu, I.; Mietta, S.; Bucca, C. Increased oral nitric oxide in obstructive sleep apnoea. Respir. Med. 2010, 104, 316–320.
- Feng, X.; Guo, X.; Lin, J.; Zhao, Z.; Tong, Z. Risk factors and fraction of exhaled nitric oxide in obstructive sleep apnea in adults. J. Int. Med. Res. 2020, 48, 0300060520926010.
- Zhang, D.; Luo, J.; Qiao, Y.; Xiao, Y.; Huang, R.; Zhong, X. Measurement of exhaled nitric oxide concentration in patients with obstructive sleep apnea: A meta-analysis. Medicine 2017, 96, e6429.
- Tie, Y.X.; Fu, Y.Y.; Xu, Z.; Peng, Y. Relationship between C-reactive protein levels and obstructive sleep apnea syndrome. Genet. Mol. Res. GMR 2016, 15, 1–5.
- Motamedi, V.; Kanefsky, R.; Matsangas, P.; Mithani, S.; Jeromin, A.; Brock, M.S.; Mysliwiec, V.; Gill, J. Elevated tau and interleukin-6 concentrations in adults with obstructive sleep apnea. Sleep Med. 2018, 43, 71–76.
- Imagawa, S.; Yamaguchi, Y.; Ogawa, K.; Obara, N.; Suzuki, N.; Yamamoto, M.; Nagasawa, T. Interleukin-6 and tumor necrosis factor-alpha in patients with obstructive sleep apnea-hypopnea syndrome. Respir. Int. Rev. Thorac. Dis. 2004, 71, 24–29.
- Fornadi, K.; Lindner, A.; Czira, M.E.; Szentkiralyi, A.; Lazar, A.S.; Zoller, R.; Turanyi, C.Z.; Veber, O.; Novak, M.; Mucsi, I.; et al. Lack of association between objectively assessed sleep disorders and inflammatory markers among kidney transplant recipients. Int. Urol. Nephrol. 2012, 44, 607–617.
- Arnardottir, E.S.; Maislin, G.; Schwab, R.J.; Staley, B.; Benediktsdottir, B.; Olafsson, I.; Juliusson, S.; Romer, M.; Gislason, T.; Pack, A.I. The interaction of obstructive sleep apnea and obesity on the inflammatory markers C-reactive protein and interleukin-6: The Icelandic Sleep Apnea Cohort. Sleep 2012, 35, 921–932.
- Sozer, V.; Kutnu, M.; Atahan, E.; Calıskaner Ozturk, B.; Hysi, E.; Cabuk, C.; Musellim, B.; Simsek, G.; Uzun, H. Changes in inflammatory mediators as a result of intermittent hypoxia in obstructive sleep apnea syndrome. Clin. Respir. J. 2018, 12, 1615–1622.
- Sinden, N.J.; Stockley, R.A. Systemic inflammation and comorbidity in COPD: A result of ‘overspill’ of inflammatory mediators from the lungs? Review of the evidence. Thorax 2010, 65, 930–936.
- Pillar, G.; Shehadeh, N. Abdominal fat and sleep apnea: The chicken or the egg? Diabetes Care 2008, 31 (Suppl. S2), S303–S309.
- Erridge, S.; Moussa, O.; McIntyre, C.; Hariri, A.; Tolley, N.; Kotecha, B.; Purkayastha, S. Obstructive Sleep Apnea in Obese Patients: A UK Population Analysis. Obes. Surg. 2021, 31, 1986–1993.
- Peppard, P.E.; Young, T.; Barnet, J.H.; Palta, M.; Hagen, E.W.; Hla, K.M. Increased prevalence of sleep-disordered breathing in adults. Am. J. Epidemiol. 2013, 177, 1006–1014.
- Di Palmo, E.; Filice, E.; Cavallo, A.; Caffarelli, C.; Maltoni, G.; Miniaci, A.; Ricci, G.; Pession, A. Childhood Obesity and Respiratory Diseases: Which Link? Children 2021, 8, 177.
- Tishler, P.V.; Larkin, E.K.; Schluchter, M.D.; Redline, S. Incidence of sleep-disordered breathing in an urban adult population: The relative importance of risk factors in the development of sleep-disordered breathing. JAMA 2003, 289, 2230–2237.
- Newman, A.B.; Foster, G.; Givelber, R.; Nieto, F.J.; Redline, S.; Young, T. Progression and regression of sleep-disordered breathing with changes in weight: The Sleep Heart Health Study. Arch. Intern. Med. 2005, 165, 2408–2413.
- Kim, A.M.; Keenan, B.T.; Jackson, N.; Chan, E.L.; Staley, B.; Poptani, H.; Torigian, D.A.; Pack, A.I.; Schwab, R.J. Tongue fat and its relationship to obstructive sleep apnea. Sleep 2014, 37, 1639–1648.
- Li, Y.; Lin, N.; Ye, J.; Chang, Q.; Han, D.; Sperry, A. Upper airway fat tissue distribution in subjects with obstructive sleep apnea and its effect on retropalatal mechanical loads. Respir. Care 2012, 57, 1098–1105.
- Stadler, D.L.; McEvoy, R.D.; Sprecher, K.E.; Thomson, K.J.; Ryan, M.K.; Thompson, C.C.; Catcheside, P.G. Abdominal compression increases upper airway collapsibility during sleep in obese male obstructive sleep apnea patients. Sleep 2009, 32, 1579–1587.
- Peppard, P.E.; Ward, N.R.; Morrell, M.J. The impact of obesity on oxygen desaturation during sleep-disordered breathing. Am. J. Respir. Crit. Care Med. 2009, 180, 788–793.
- Phillips, B.G.; Hisel, T.M.; Kato, M.; Pesek, C.A.; Dyken, M.E.; Narkiewicz, K.; Somers, V.K. Recent weight gain in patients with newly diagnosed obstructive sleep apnea. J. Hypertens. 1999, 17, 1297–1300.
- Kuvat, N.; Tanriverdi, H.; Armutcu, F. The relationship between obstructive sleep apnea syndrome and obesity: A new perspective on the pathogenesis in terms of organ crosstalk. Clin. Respir. J. 2020, 14, 595–604.
- Ong, C.W.; O’Driscoll, D.M.; Truby, H.; Naughton, M.T.; Hamilton, G.S. The reciprocal interaction between obesity and obstructive sleep apnoea. Sleep Med. Rev. 2013, 17, 123–131.
- Hoyos, C.M.; Murugan, S.M.; Melehan, K.L.; Yee, B.J.; Phillips, C.L.; Killick, R.; Cayanan, E.A.; Wong, K.K.; Liu, P.Y.; Grunstein, R.R.; et al. Dose-dependent effects of continuous positive airway pressure for sleep apnea on weight or metabolic function: Individual patient-level clinical trial meta-analysis. J. Sleep Res. 2019, 28, e12788.
- Ou, Q.; Chen, B.; Loffler, K.A.; Luo, Y.; Zhang, X.; Chen, R.; Wang, Q.; Drager, L.F.; Lorenzi-Filho, G.; Hlavac, M.; et al. The Effects of Long-term CPAP on Weight Change in Patients with Comorbid OSA and Cardiovascular Disease: Data From the SAVE Trial. Chest 2019, 155, 720–729.
- De Luca Canto, G.; Pachêco-Pereira, C.; Aydinoz, S.; Major, P.W.; Flores-Mir, C.; Gozal, D. Biomarkers associated with obstructive sleep apnea and morbidities: A scoping review. Sleep Med. 2015, 16, 347–357.
- Ryan, S.; Arnaud, C.; Fitzpatrick, S.F.; Gaucher, J.; Tamisier, R.; Pépin, J.L. Adipose tissue as a key player in obstructive sleep apnoea. Eur. Respir. Rev. Off. J. Eur. Respir. Soc. 2019, 28, 190006.
- Huang, Y.S.; Guilleminault, C.; Hwang, F.M.; Cheng, C.; Lin, C.H.; Li, H.Y.; Lee, L.A. Inflammatory cytokines in pediatric obstructive sleep apnea. Medicine 2016, 95, e4944.
- Bhatt, S.P.; Guleria, R.; Kabra, S.K. Metabolic alterations and systemic inflammation in overweight/obese children with obstructive sleep apnea. PLoS ONE 2021, 16, e0252353.
- Tauman, R.; Serpero, L.D.; Capdevila, O.S.; O’Brien, L.M.; Goldbart, A.D.; Kheirandish-Gozal, L.; Gozal, D. Adipokines in children with sleep disordered breathing. Sleep 2007, 30, 443–449.
- Söğüt, A.; Açıkgöz, Ş.; Uzun, L.; Uğur, M.B.; Altın, R.; Dağlı, E.; Kaditis, A.; Ersu, R. Leptin levels in children with obstructive sleep apnea syndrome. Tuberk. Toraks 2016, 64, 283–288.
- Yang, H.; Engeland, C.G.; King, T.S.; Sawyer, A.M. The relationship between diurnal variation of cytokines and symptom expression in mild obstructive sleep apnea. J. Clin. Sleep Med. JCSM Off. Publ. Am. Acad. Sleep Med. 2020, 16, 715–723.
- Semenza, G.L. Hypoxia-inducible factors in physiology and medicine. Cell 2012, 148, 399–408.
- Kaelin, W.G., Jr.; Ratcliffe, P.J. Oxygen sensing by metazoans: The central role of the HIF hydroxylase pathway. Mol. Cell 2008, 30, 393–402.
- Epstein, A.C.; Gleadle, J.M.; McNeill, L.A.; Hewitson, K.S.; O’Rourke, J.; Mole, D.R.; Mukherji, M.; Metzen, E.; Wilson, M.I.; Dhanda, A.; et al. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell 2001, 107, 43–54.
- Manalo, D.J.; Rowan, A.; Lavoie, T.; Natarajan, L.; Kelly, B.D.; Ye, S.Q.; Garcia, J.G.; Semenza, G.L. Transcriptional regulation of vascular endothelial cell responses to hypoxia by HIF-1. Blood 2005, 105, 659–669.
- Drager, L.F.; Jun, J.C.; Polotsky, V.Y. Metabolic consequences of intermittent hypoxia: Relevance to obstructive sleep apnea. Best Pract. Res. Clin. Endocrinol. Metab. 2010, 24, 843–851.
- Weidemann, A.; Johnson, R.S. Biology of HIF-1alpha. Cell Death Differ. 2008, 15, 621–627.
- Jhamb, M.; Unruh, M. Bidirectional relationship of hypertension with obstructive sleep apnea. Curr. Opin. Pulm. Med. 2014, 20, 558–564.
- Hsu, H.C.; Chen, N.H.; Ho, W.J.; Lin, M.H. Factors associated with undiagnosed obstructive sleep apnoea among hypertensive patients: A multisite cross-sectional survey study in Taiwan. J. Clin. Nurs. 2018, 27, 1901–1912.
- Bouloukaki, I.; Grote, L.; McNicholas, W.T.; Hedner, J.; Verbraecken, J.; Parati, G.; Lombardi, C.; Basoglu, O.K.; Pataka, A.; Marrone, O.; et al. Mild obstructive sleep apnea increases hypertension risk, challenging traditional severity classification. J. Clin. Sleep Med. JCSM Off. Publ. Am. Acad. Sleep Med. 2020, 16, 889–898.
- Cai, A.; Wang, L.; Zhou, Y. Hypertension and obstructive sleep apnea. Hypertens. Res. Off. J. Jpn. Soc. Hypertens. 2016, 39, 391–395.
- Lavie, P.; Herer, P.; Hoffstein, V. Obstructive sleep apnoea syndrome as a risk factor for hypertension: Population study. BMJ Clin. Res. Ed. 2000, 320, 479–482.
- Wang, Y.; Li, C.; Feng, L.; Feng, J.; Cao, J.; Chen, B. Prevalence of hypertension and circadian blood pressure variations in patients with obstructive sleep apnoea-hypopnoea syndrome. J. Int. Med. Res. 2014, 42, 773–780.
- Volná, J.; Kemlink, D.; Kalousová, M.; Vávrová, J.; Majerová, V.; Mestek, O.; Svarcová, J.; Sonka, K.; Zima, T. Biochemical oxidative stress-related markers in patients with obstructive sleep apnea. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2011, 17, Cr491–Cr497.
- Olszewska, E.; Pietrewicz, T.M.; Świderska, M.; Jamiołkowski, J.; Chabowski, A. A Case-Control Study on the Changes in High-Sensitivity C-Reactive Protein and Tumor Necrosis Factor-Alpha Levels with Surgical Treatment of OSAS. Int. J. Mol. Sci. 2022, 23, 14116.
- Takahashi, K.; Chin, K.; Nakamura, H.; Morita, S.; Sumi, K.; Oga, T.; Matsumoto, H.; Niimi, A.; Fukuhara, S.; Yodoi, J.; et al. Plasma thioredoxin, a novel oxidative stress marker, in patients with obstructive sleep apnea before and after nasal continuous positive airway pressure. Antioxid. Redox Signal. 2008, 10, 715–726.
- Guo, Q.; Wang, Y.; Li, Q.Y.; Li, M.; Wan, H.Y. Levels of thioredoxin are related to the severity of obstructive sleep apnea: Based on oxidative stress concept. Sleep Breath. Schlaf Atm. 2013, 17, 311–316.
- Jordan, W.; Cohrs, S.; Degner, D.; Meier, A.; Rodenbeck, A.; Mayer, G.; Pilz, J.; Rüther, E.; Kornhuber, J.; Bleich, S. Evaluation of oxidative stress measurements in obstructive sleep apnea syndrome. J. Neural Transm. 2006, 113, 239–254.
- Cofta, S.; Wysocka, E.; Piorunek, T.; Rzymkowska, M.; Batura-Gabryel, H.; Torlinski, L. Oxidative stress markers in the blood of persons with different stages of obstructive sleep apnea syndrome. J. Physiol. Pharmacol. Off. J. Pol. Physiol. Soc. 2008, 59 (Suppl. S6), 183–190.
- Pau, M.C.; Zinellu, E.; Fois, S.S.; Piras, B.; Pintus, G.; Carru, C.; Mangoni, A.A.; Fois, A.G.; Zinellu, A.; Pirina, P. Circulating Malondialdehyde Concentrations in Obstructive Sleep Apnea (OSA): A Systematic Review and Meta-Analysis with Meta-Regression. Antioxidants 2021, 10, 1053.
- Kizawa, T.; Nakamura, Y.; Takahashi, S.; Sakurai, S.; Yamauchi, K.; Inoue, H. Pathogenic role of angiotensin II and oxidised LDL in obstructive sleep apnoea. Eur. Respir. J. 2009, 34, 1390–1398.
- Fadaei, R.; Safari-Faramani, R.; Rezaei, M.; Ahmadi, R.; Rostampour, M.; Moradi, N.; Khazaie, H. Circulating levels of oxidized low-density lipoprotein in patients with obstructive sleep apnea: A systematic review and meta-analysis. Sleep Breath. Schlaf Atm. 2020, 24, 809–815.
- Mancuso, M.; Bonanni, E.; LoGerfo, A.; Orsucci, D.; Maestri, M.; Chico, L.; DiCoscio, E.; Fabbrini, M.; Siciliano, G.; Murri, L. Oxidative stress biomarkers in patients with untreated obstructive sleep apnea syndrome. Sleep Med. 2012, 13, 632–636.
- Simiakakis, M.; Kapsimalis, F.; Chaligiannis, E.; Loukides, S.; Sitaras, N.; Alchanatis, M. Lack of effect of sleep apnea on oxidative stress in obstructive sleep apnea syndrome (OSAS) patients. PLoS ONE 2012, 7, e39172.
- Alzoghaibi, M.A.; Bahammam, A.S. Lipid peroxides, superoxide dismutase and circulating IL-8 and GCP-2 in patients with severe obstructive sleep apnea: A pilot study. Sleep Breath. Schlaf Atm. 2005, 9, 119–126.
- Ntalapascha, M.; Makris, D.; Kyparos, A.; Tsilioni, I.; Kostikas, K.; Gourgoulianis, K.; Kouretas, D.; Zakynthinos, E. Oxidative stress in patients with obstructive sleep apnea syndrome. Sleep Breath. Schlaf Atm. 2013, 17, 549–555.
- Jullian-Desayes, I.; Joyeux-Faure, M.; Tamisier, R.; Launois, S.; Borel, A.L.; Levy, P.; Pepin, J.L. Impact of obstructive sleep apnea treatment by continuous positive airway pressure on cardiometabolic biomarkers: A systematic review from sham CPAP randomized controlled trials. Sleep Med. Rev. 2015, 21, 23–38.
- Paz, Y.M.H.L.; Hazen, S.L.; Tracy, R.P.; Strohl, K.P.; Auckley, D.; Bena, J.; Wang, L.; Walia, H.K.; Patel, S.R.; Mehra, R. Effect of Continuous Positive Airway Pressure on Cardiovascular Biomarkers: The Sleep Apnea Stress Randomized Controlled Trial. Chest 2016, 150, 80–90.
- Lira, A.B.; De Sousa Rodrigues, C.F. Evaluation of oxidative stress markers in obstructive sleep apnea syndrome and additional antioxidant therapy: A review article. Sleep Breath. Schlaf Atm. 2016, 20, 1155–1160.
- Chaiard, J.; Bhatarasakoon, P. Effectiveness of behavioral and psychosocial interventions for continuous positive airway pressure adherence among patients with obstructive sleep apnea: A systematic review and meta-analysis. Appl. Nurs. Res. 2023, 69, 151654.
- Voulgaris, A.; Archontogeorgis, K.; Anevlavis, S.; Fanaridis, M.; Froudarakis, M.E.; Schiza, S.; Steiropoulos, P. Effect of compliance to continuous positive airway pressure on exacerbations, lung function and symptoms in patients with chronic obstructive pulmonary disease and obstructive sleep apnea (overlap syndrome). Clin. Respir. J. 2023, 17, 165–175.
- Grebe, M.; Eisele, H.J.; Weissmann, N.; Schaefer, C.; Tillmanns, H.; Seeger, W.; Schulz, R. Antioxidant vitamin C improves endothelial function in obstructive sleep apnea. Am. J. Respir. Crit. Care Med. 2006, 173, 897–901.
- Celec, P.; Jurkovičová, I.; Buchta, R.; Bartík, I.; Gardlík, R.; Pálffy, R.; Mucska, I.; Hodosy, J. Antioxidant vitamins prevent oxidative and carbonyl stress in an animal model of obstructive sleep apnea. Sleep Breath. Schlaf Atm. 2013, 17, 867–871.
- Jin, Z.N.; Wei, Y.X. Meta-analysis of effects of obstructive sleep apnea on the renin-angiotensin-aldosterone system. J. Geriatr. Cardiol. 2016, 13, 333–343.
- Hansson, G.K. Inflammation, atherosclerosis, and coronary artery disease. N. Engl. J. Med. 2005, 352, 1685–1695.
- Gileles-Hillel, A.; Almendros, I.; Khalyfa, A.; Zhang, S.X.; Wang, Y.; Gozal, D. Early intermittent hypoxia induces proatherogenic changes in aortic wall macrophages in a murine model of obstructive sleep apnea. Am. J. Respir. Crit. Care Med. 2014, 190, 958–961.
- Castro-Grattoni, A.L.; Alvarez-Buvé, R.; Torres, M.; Farré, R.; Montserrat, J.M.; Dalmases, M.; Almendros, I.; Barbé, F.; Sánchez-de-la-Torre, M. Intermittent Hypoxia-Induced Cardiovascular Remodeling Is Reversed by Normoxia in a Mouse Model of Sleep Apnea. Chest 2016, 149, 1400–1408.
- Díaz-García, E.; García-Tovar, S.; Alfaro, E.; Jaureguizar, A.; Casitas, R.; Sánchez-Sánchez, B.; Zamarrón, E.; Fernández-Lahera, J.; López-Collazo, E.; Cubillos-Zapata, C.; et al. Inflammasome Activation: A Keystone of Proinflammatory Response in Obstructive Sleep Apnea. Am. J. Respir. Crit. Care Med. 2022, 205, 1337–1348.
- Jelic, S.; Padeletti, M.; Kawut, S.M.; Higgins, C.; Canfield, S.M.; Onat, D.; Colombo, P.C.; Basner, R.C.; Factor, P.; LeJemtel, T.H. Inflammation, oxidative stress, and repair capacity of the vascular endothelium in obstructive sleep apnea. Circulation 2008, 117, 2270–2278.
- Drager, L.F.; Yao, Q.; Hernandez, K.L.; Shin, M.K.; Bevans-Fonti, S.; Gay, J.; Sussan, T.E.; Jun, J.C.; Myers, A.C.; Olivecrona, G.; et al. Chronic intermittent hypoxia induces atherosclerosis via activation of adipose angiopoietin-like 4. Am. J. Respir. Crit. Care Med. 2013, 188, 240–248.
More