Liver cirrhosis is a chronic disease that can be complicated by episodes of decompensation such as variceal bleeding, hepatic encephalopathy, ascites, and jaundice, with subsequent increased mortality. Infections are also among the most common complications in cirrhotic patients, mostly due to a defect in immunosurveillance. Among them, one of the most frequent is spontaneous bacterial peritonitis (SBP), defined as the primary infection of ascitic fluid without other abdominal foci. SBP is mainly induced by Gram-negative bacteria living in the intestinal tract, and translocating through the intestinal barrier, which in cirrhotic patients is defective and more permeable.
- cirrhosis
- spontaneous bacterial peritonitis
- gut-liver axis
- microbiota
- intestinal barrier
- bacterial translocation
1. Introduction
2. Infections and Liver Cirrhosis
Infections are a major cause of cirrhosis decompensation and globally increase mortality fourfold [10][11]. Approximately two-thirds of patients with cirrhosis and extrahepatic organ failure present with sepsis; in-hospital mortality in cirrhotic patients with severe sepsis exceeds 75%, and the risk of sepsis-related death due to multi-drug resistant bacteria is currently increasing as new therapies for such microorganisms are still not available, representing an unsolved public health concern [12][13]. The higher infectious risk of cirrhotic patients could be related to specific molecular and cellular mechanisms associated with liver dysfunction, but also with other host-related factors (e.g., old age, malnutrition, immunosuppressive drugs use) and with the gut microbiota. So far, the balanced interaction between the host and the gut microbiota has been considered a protective factor against all infection types in cirrhotic patients. In fact, gut microbiota is critical in providing resistance against colonization by exogenous microorganisms, and dysbiosis has been associated with a higher risk of infection [14]. Gut microbiota analysis performed on cirrhotic patients with nosocomial infections showed a reduction in commensal bacteria and an increase in pathogens [15]. Therefore, immune system, intestinal microenvironment, and host factors are the main actors in modulating the risk of infections in cirrhotic patients.2.1. Immune-Paralysis: Molecular and Cellular Mechanisms
Decompensated cirrhosis is usually characterized by a multi-systemic inflammatory condition, in which immune innate cells are constantly stimulated by pro-inflammatory molecules, such as pathogens-associated molecular patterns (PAMPs) derived from bacteria translocated through the intestinal mucosa. PAMPs trigger the local inflammatory response inducing cellular death, with the consequent release of damage-associated molecular patterns (DAMPs), which, interacting with immune cells, enhance inflammation. On the other side, the immune system sets in place a condition of tolerance, which is known as endotoxin tolerance [16]. Emerging data reported that during bacterial infection associated with decompensated cirrhosis, certain types of monocytes, known as monocytic myeloid-derived suppressor cells (M-MDSCs) and MER-receptor tyrosine kinase (MERTK) cells, migrate to the infection site impairing the potential activation of T cells; indeed, the phenotype of effector CD8+ T cells changes into the exhausted one, which is characterized by increased expression of programmed cell death protein 1 (PD-1) and T cell immunoglobulin and mucin domain-containing protein 3 (TIM-3) [17][18]. In addition, it has been recently demonstrated that liver cirrhosis decompensation is associated with increased blood levels of both soluble stimulating and inhibitory immune checkpoint receptors. Both the immune-stimulating sCD40 receptor and immune-suppressive soluble B and T-cell lymphocyte-associated (sBTLA) receptor seem to be involved, highlighting the ambivalent role of lymphocytes that could change from an effector into an exhausted phenotype [19]. Furthermore, cirrhosis impairs the hepatic reticuloendothelial system, by altering Kupffer cells’ function, inducing sinusoidal capillarization, and resulting in the recruitment of dysfunctional immune cells [20]. In patients with cirrhosis, hepatic synthesis of complement proteins and acute-phase proteins decreases, leading to the impairment of opsonization and the reduction of chemotactic activity, as well as of macrophages phagocytic activity . Large CD14+ CD206+ peritoneal macrophages are another specific subpopulation of immunosuppressive monocytes, which is typical of decompensated cirrhosis. This macrophage population is responsible for the dysregulation of the innate immunity in the ascitic fluid [20]. Recent evidence has demonstrated that during SBP, large peritoneal macrophages secreted a cleaved protein named soluble mannose receptor (sCD206), which is associated with an increased local and systemic inflammatory response [21]. CD206 is typically expressed by M2 macrophages, an anti-inflammatory phenotype of polarized macrophages [22][23]. Several phenotypes of macrophages have been described; among them, M1 and M2 are the most well-known. M1 macrophages principally produce pro-inflammatory cytokines and seem to be mostly involved in acute liver injury [24]. On the other hand, M2 polarization is more related to liver fibrosis/cirrhosis progression [25] and development of complications, such as hepatocellular carcinoma (HCC) [26]. Although the mechanisms driving the M2 polarization are complex and not completely understood, the gut microbiota seems to play a significant role. Dysbiosis can directly induce an M2 phenotype through the interaction between PAMPs and toll-like receptor 4 (TLR4) expressed on macrophage surface [27]. Nonetheless, a balanced macrophage polarization is essential for the organism response to bacterial infections [28]. On the contrary, the loss of this complex equilibrium can lead to a higher rate of infectious disease and is associated with a worse prognosis. Increased blood levels of sCD206 have been proposed as a marker of abnormal M2 polarization and have been related to the severity of some infections, such as community-acquired pneumonia [29]. As a consequence, inflammation is constantly activated but immune cells lose their physiological effectiveness, becoming unable to face pathogenic stimulations and to maintain a homeostatic interaction between the intestinal bacteria and the host. This inability of the immune system seems to be crucially involved in the increased susceptibility to infections of patients with cirrhosis. Due to this immune dysfunction, an apparent unresponsiveness against infections and cellular damage occurs, in a condition known as immune-paralysis [30]. Furthermore, the persistent overflow of PAMPs and their translocation in the systemic circulation contributes to worsening the pre-existing hyperdynamic circulation associated with cirrhosis, leading to uncontrolled systemic vasodilation, impaired cardiac output, and hypotension, with a compensatory hyperactivation of the renin–angiotensin–aldosterone system that also precipitates cirrhosis decompensation [31]. In an attempt to limit infection and inflammation spreading, when sepsis occurs, patients with acute on chronic liver failure (ACLF), IL6, IL17, and IL10 are over-produced, further worsening liver cellular damage and coagulation dysfunction [32].2.2. Infections and Liver Cirrhosis: Clinical Concerns
About 25–46% of cirrhotic patients are hospitalized for acute decompensation due to bacterial infections, two-third of whom diagnosed at admission (within 48 h): 30–50% are community-acquired, 25–40% are health care-associated, whereas one-third are nosocomial. Even though SBP has been reported to be the most common type of infection, with a prevalence of 25–35%, sites of extra-peritoneal colonization could also lead to life-threatening complications. Among them, 20–25% are represented by urinary tract infections (UTIs), 20% by pneumonia, 8–15% by spontaneous bloodstream infections, and 5–10% by skin and soft tissues infections (SSTIs) [33] (Figure 1). Nosocomial infections often develop after invasive procedures, as central venous and urinary catheters placement or mechanical ventilation, as the disruption of innate immune barriers realizes an entrance gate into the bloodstream for pathogens and commensal bacteria [30]. About half of all infections recognize a bacterial cause, with a prevalence of Gram-negative over Gram-positive strains, while 10–15% of patients suffer from secondary fungal infections [33]. High mortality has also been observed among patients with cirrhosis who have severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection [34]. Enterobacteriaceae (such as Escherichia coli and Klebsiella pneumoniae) and other gram-negative bacteria are the main pathogens in SBP and UTIs (50–70%), with a minor contribution of Gram-positive bacteria as Staphylococcus aureus and Enterococci (30–45%). On the contrary, pneumonia and SSTIs are more frequently caused by Gram-positive strains. Due to the high prevalence of health care-associated infections, involved bacteria are frequently multi-drug resistant (MDR), as extended spectrum beta lactamase and AmpC-producing Enterobacteriaceae, methicillin-resistant Staphylococcus aureus, and vancomycin-resistant Enterococci, or extensively drug-resistant (XDR), as carbapenemase-producing Enterobacteriaceae, carbapenem-resistant Pseudomonas aeruginosa, and Acinetobacter baumannii [33]. In the last two decades, the proportion of resistant bacteria has increased due to a widespread use of quinolones for SBP prophylaxis and of third-generation cephalosporines for treatment of other nosocomial infections; this represents an important challenge for clinicians, as the risk of antibiotic failure, progression to septic shock, and overall hospital mortality is increased [35].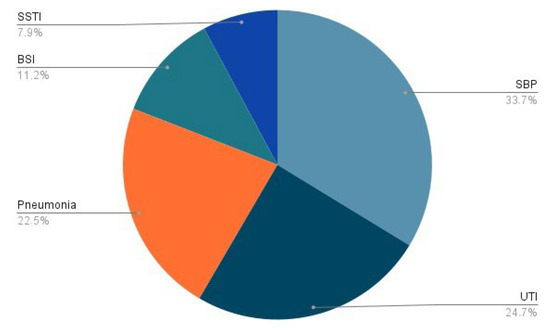
2.3. Clinical Features of SBP
SBP is the main cause of infection in individuals with liver cirrhosis, occurring in 25–35% of decompensated cirrhotic inpatients and in 3.5% of decompensated cirrhotic outpatients, with a survival rate per year of 30–50%. SBP diagnosis is based on the finding of an ascitic polymorphonuclear cell count ≥ 250/microL, with either positive or negative microbiological culture. Aerobic Gram-negative bacteria, such as Escherichia coli and Klebsiella pneumoniae, are those most frequently implicated [36]. Immunological dysfunction is strongly involved in SBP pathogenesis, and comorbidities can contribute to the immunosuppression promoting SBP onset. Malnutrition is frequent in patients with cirrhosis and has been associated with higher prevalence of SPB [37]. Indeed, lower levels of serum zinc are an independent predictor of hepatic decompensation and SBP [38]. Vitamin D deficiency seems also to be related with an increased risk of SBP in patients with HCV-related cirrhosis [39]. Furthermore, lower serum levels of 25-hydroxyvitamin D could predict a worse outcome in these patients [39]. Other systemic comorbidities usually associated with an immune dysregulation are implicated in SBP development, such as type 2 diabetes, which increases the risk of SBP six-fold in patients with cirrhosis [40]. However, SBP is characterized by the absence of a primary source of intra-abdominal infection. For this reason, alongside the immune system dysregulation, the loss of intestinal mucosal barrier and the consequent gut bacteria translocation are considered to be the main pathogenic mechanism, thus confirming a crucial role of the gut–liver axis.3. The Gut–Liver Axis in the Pathogenesis of SBP
3.1. Bacterial Translocation in Liver Cirrhosis
Portal hypertension causes a profound dysfunction of the gut–liver axis. The vasodilation of splanchnic vessels reduces blood flow velocity and capacitance in the intestinal mucosa, leading to a chronic ischemic condition. This acts as an inflammatory trigger, inducing fibromuscular proliferation of the intestinal layers and modifying the morphological characteristics of enterocytes [41]. In a descriptive pilot study, intestinal biopsies from a population of cirrhotic patients showed a reduced representation of tight junctions (TJ) proteins such as occludin and claudin 1. Histologically, loss of microvilli and reduced villi to crypt ratio have been evidenced too [41][42]. As a result of the epithelial barrier dysfunction, intestinal permeability is increased, realizing a condition also known as leaky gut, which promotes bacterial translocation and a consequent inflammatory cascade (Figure 2) [41]. The release of small amounts of PAMPs, such as endotoxins, lipopolysaccharides (LPS), and bacterial DNA, from the gut into the portal venous system to extra-intestinal sites is a physiological process, which has been described even in healthy subjects, with no clinical consequences. However, in patients with liver cirrhosis, bacterial translocation is exacerbated by several pathological mechanisms such as reduced secretion of defensins and regenerating islet-derived proteins (RegIIIβ and RegIIIγ), thickening of the inner mucus layer by over-expression of MUC2, reduced intestinal intraluminal concentration of secondary bile acids, decreased mucosal IgA secretion, impaired gastric acid secretion, and slower oro-cecal transit time (OCTT) due to ineffective gastrointestinal peristalsis [43]. Bacterial translocation underlies immunotolerance in cirrhotic patients; indeed, chronic inflammation induces a dysfunction of the innate and adaptive immune response, which together with the reduced number of circulating leukocytes due to hypersplenism and the reduced production of humoral and complement factor leads to a sort of “immune paralysis” [44]. Moreover, phagocytosis stimulated by the binding of LPS with TLR4 on the macrophage surface is ineffective, and secondly the Kupffer cells activated by bacterial products release multiple cytokines in a sort of “cytokine storm”, enhancing liver damage [45]. The vicious circle that links gut dysbiosis, bacterial translocation, and liver cirrhosis is described in Figure 2.
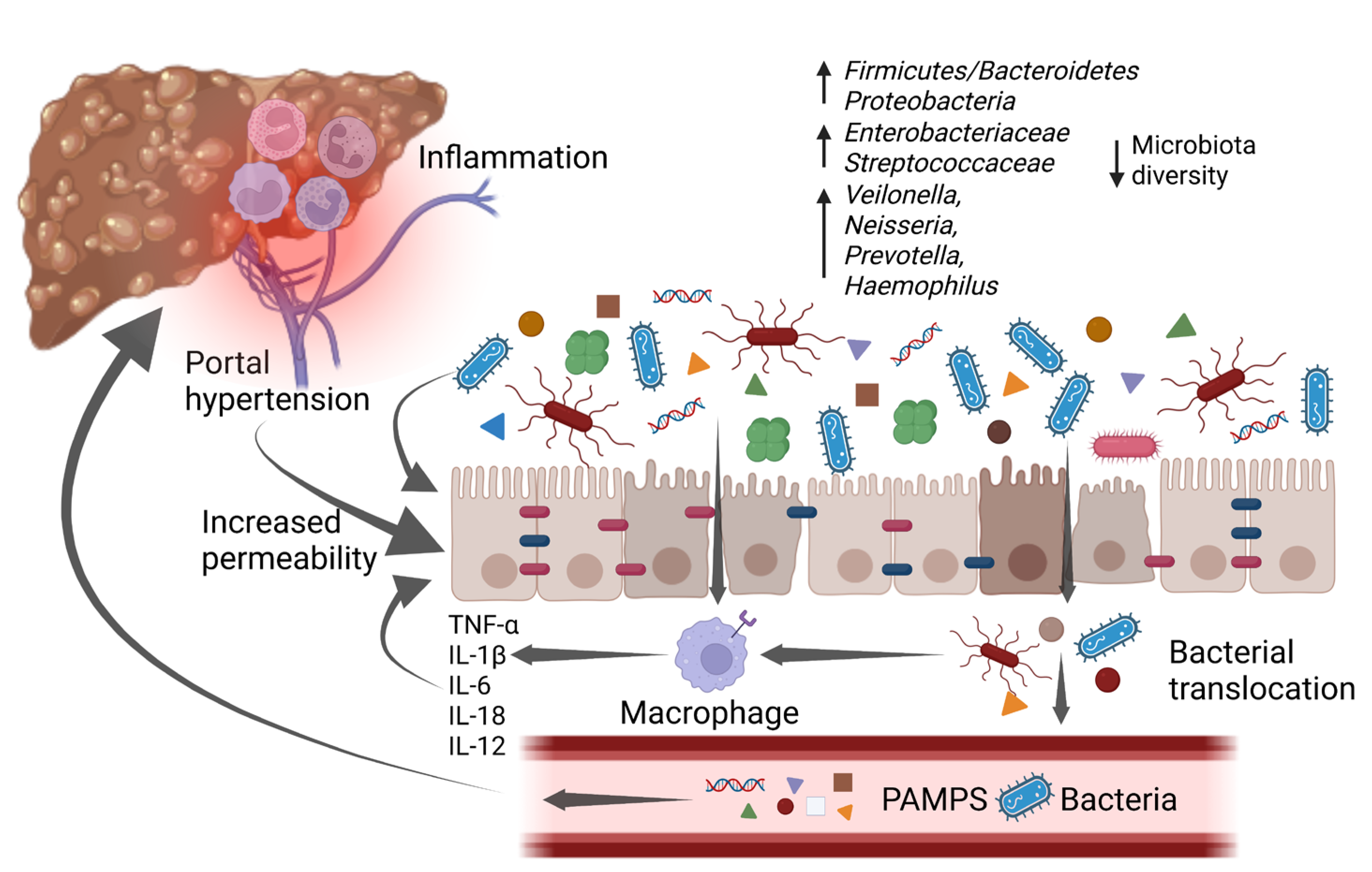
![]() Figure 2. An increase in pro-inflammatory bacteria and a reduction in overall diversity are typical of gut microbiota changes associated with cirrhosis. These changes promote a low-grade chronic inflammatory state that impairs intestinal barrier function, which is already affected by portal hypertension, leading to a condition known as “leaky gut”. The increased intestinal permeability, along with the disrupted immune system, allows bacterial translocation and the increase of bacterial metabolites in the bloodstream. As a consequence, it enhances inflammation which worsens intestinal barrier disruption and in the liver promotes cirrhosis progression, with the emergence of its complications. IL-1β: interleukin-1β; IL-6: interleukin-6; IL-12: interleukin-12; IL-18: interleukin-18; PAMPs: pathogen-associated molecular patterns; TNF-α: tumor necrosis factor α. Created with BioRender.com accessed on 10 January 2023.
Figure 2. An increase in pro-inflammatory bacteria and a reduction in overall diversity are typical of gut microbiota changes associated with cirrhosis. These changes promote a low-grade chronic inflammatory state that impairs intestinal barrier function, which is already affected by portal hypertension, leading to a condition known as “leaky gut”. The increased intestinal permeability, along with the disrupted immune system, allows bacterial translocation and the increase of bacterial metabolites in the bloodstream. As a consequence, it enhances inflammation which worsens intestinal barrier disruption and in the liver promotes cirrhosis progression, with the emergence of its complications. IL-1β: interleukin-1β; IL-6: interleukin-6; IL-12: interleukin-12; IL-18: interleukin-18; PAMPs: pathogen-associated molecular patterns; TNF-α: tumor necrosis factor α. Created with BioRender.com accessed on 10 January 2023.
3.2. The Gut Microbiota in Cirrhotic Patients with SBP
Cirrhotic patients typically present qualitative and quantitative alterations of the gut microbiota composition, which worsen with the progression of the disease and hamper intestinal permeability and bacterial translocation [31][46]. Much evidence suggests that cirrhosis-related dysbiosis is characterized by the overabundance of pathogens such as Enterobacteriaceae and the depletion of beneficial commensals; furthermore, it is associated with the development of decompensation episodes as well as HCC, and may even identify patients at unfavorable prognosis [47][48][49][50]. Bacterial DNA can be detected in the ascites of half of cirrhotic patients even in the absence of SBP, probably due to bacterial opsonization [44]. Enterobacteriaceae, Streptococcus pneumoniae, and Streptococcus viridans are the bacterial species most frequently detected in the ascitic fluid of patients with SBP; among them, Escherichia coli is reported to be the most abundant, but also Gram-positive bacteria such as Staphylococcus, Streptococcus, and Enterococcus, as well as Helicobacter spp., including Helicobacter pylori, have been described [51]. Indeed, a remarkable result of the gastrointestinal dysfunction associated with liver cirrhosis is the oralization of the small intestinal and the colonic microbiota. About 48–73% of cirrhotic patients present a small intestinal bacterial overgrowth (SIBO), especially those with previous episodes of SBP or hepatic encephalopathy [52]. In particular, oral species such as Streptococcus oralis and Streptococcus parasanguinis not only co-existed with enteric species, such as Enterococcus spp., Clostridium spp., and Erysipelatoclostridium ramnosum, but were correlated with disease severity [48].
Nonetheless, SBP is associated with further gut microbiota perturbations. In fact, a lower alpha and beta diversity was observed in the gut microbiota of patients with SBP, with an increase of Pantoea, Serratia marcescens, Klebsiella pneumoniae, Prevotella oris, Escherichia coli, Clostridioides difficile, Enterococcus, Veillonella spp., and other potential pathobionts, as well as a lower abundance of the Ruminococcus torques, Faecalibacterium prausnitzii, Methanobrevibacter smithii, and Lactobacillus reuteri [53][54].
4. Gut Microbiota Modulation as a Future Treatment for SBP
Since the gut microbiota plays a pivotal role in the onset and the progression of decompensated cirrhosis, its modulation might be used as a modifier of the natural course of the disease. Available data on the gut microbiota modulation of antibiotic SBP treatment show that norfloxacin, which is the principle antibiotic used in SBP prophylaxis, can modulate gut microbiota. Indeed, norfloxacin prophylaxis has been associated with a significant decrease of Enterobacteriaceae, reaching undetectable levels [55]. As a matter of fact, SBP prophylaxis increased the relative abundance of Gram-positive bacteria, mostly due to the elimination of Gram-negative species [53]. Other antibiotics can modulate gut microbiota. Rifaximin, which is a non absorbable antibiotic acting locally in the gastrointestinal tract, has shown its eubiotic properties. In fact rifaximin treatment significantly increases the abundance of gut beneficial bacteria, such as Lactobacillus spp., decreasing harmful ones (e.g. Veillonellaceae) [56][57], and also preventing the oralization of the gut microbiota [58]. As a result, rifaximin has been suggested as a possible alternative to norfloxacin in secondary SBP prophylaxis [59].4.1. Probiotics, Prebiotics and Synbiotics
It has been reported that probiotic supplementation increases resistance to enteric infections in IL10-deficient mice and reduces endotoxemia in cirrhotic patients by modulating the viable counts of potentially pathogenic Gram-positive and Gram-negative bacteria in the intestine [60]. Based on this premise, clinical studies tested the administration of Lactobacillus-containing probiotics as a primary therapy in SBP, showing conflicting results. On one hand, Lactobacillus GG was not effective in preventing bacterial translocation and SBP, whilst the administration of Lactobacillus johnsonii plus antioxidants was able to obtain a reduction in bacterial translocation [61]; on the other hand, the addition of probiotics to norfloxacin did not improve the clinical efficacy in primary or secondary prophylaxis. Thus, the current literature does not support the use of probiotics to prevent the occurrence of SBP [62]. Prebiotics and synbiotics are commonly used to modulate imbalances in the gut microbiota [63]. It has been demonstrated that both prebiotics and symbiotics can reduce bacterial translocation from the gut [64]. Prebiotics have been found to increase the amount of occludin, claudin-3, and ZO-1, thereby restoring impaired tight junctions in the intestinal epithelium [65]. Given their impact on the gut microbiota and intestinal barrier, prebiotics and synbiotics could potentially be considered as an additional prevention strategy for SBP. However, to date, neither prebiotics nor synbiotics have been investigated for this purpose, or have been included in the guidelines for SBP management [66].4.2. Enterosorbent Compounds and Bile Acids Signaling
An innovative strategy involves the absorption of intraluminal host or microbial metabolites or ligands, with the aim of reducing the translocation of pathogenic factors, such as ammonia or endotoxin, or modulating bile acid pathways [67]. Yaq-001 (Yaqrit Limited, Herefordshire, UK), a new non-absorbable synthetic carbon able to bind large molecular weight substances such as endotoxin, exotoxins, and cytokines, was found to improve liver injury, portal pressure, and LPS-induced ROS production in an in vivo model of cirrhosis [68]. Additionally, the phosphate sequestrant sevelamer is able to improve gut microbiota alpha-diversity and bind intraluminal endotoxin [69]. The administration of obethicolic acid, a farnesoid X receptor (FXR) agonist acting via bile acids signaling pathway, for 2 weeks to cirrhotic rats with ascites decreased bacterial translocation from 78.3% to 33.3%, significantly modulating mucosal microbiota composition and improving intestinal barrier function [69][70].4.3. Intestinal Motility
As the increased OCTT is considered one of the factors contributing to SIBO and bacterial translocation, the use of prokinetics such as cisapride, a 5-HT4-agonist, is supposed to reduce the incidence of SBP. An animal study analyzing the efficacy of cisapride and norfloxacin administration versus cisapride alone showed a reduction in the incidence of SBP in ascitic patients [71]. Another animal study demonstrated that rats treated with propranolol had a lower portal pressure, a shorter OCTT, and lesser occurrence of bacterial translocation and SIBO compared to rats receiving placebo, resulting in a decreased risk of SBP [72].4.4. Other Experimental Treatments
Further ongoing studies are evaluating the effectiveness of bacteriophages, carbon nanoparticles-binding microbial components, and micro-RNA (as miR-320a against Escherichia Coli) treatments in reducing dysbiosis in patients with cirrhosis-related complications, but evidence on the efficacy of such treatments on SBP is expected in the near future [73].References
- Blachier, M.; Leleu, H.; Peck-Radosavljevic, M.; Valla, D.C.; Roudot-Thoraval, F. The burden of liver disease in Europe: A review of available epidemiological data. J. Hepatol. 2013, 58, 593–608.
- Mokdad, A.A.; Lopez, A.D.; Shahraz, S.; Lozano, R.; Mokdad, A.H.; Stanaway, J. Liver cirrhosis mortality in 187 countries between 1980 and 2010: A systematic analysis. BMC Med. 2014, 12, 14.
- Yung, Y.K.; Yim, H.J. Reversal of liver cirrhosis: Current evidence and expectations. Korean J. Intern. Med. 2017, 32, 213–228.
- Tsochatzis, E.A.; Bosch, J.; Burroughs, A.K. Liver cirrhosis. Lancet 2014, 383, 1749–1761.
- Piano, S.; Tonon, M.; Angeli, P. Changes in the epidemiology and management of bacterial infections in cirrhosis. Clin. Mol. Hepatol. 2021, 27, 437–445.
- Ascione, T.; Di Flumeri, G.; Boccia, G.; De Caro, F. Infections in patients affected by liver cirrhosis: An update. Infez. Med. 2017, 25, 91–97.
- Di Tommaso, N.; Gasbarrini, A.; Ponziani, F.R. Intestinal Barrier in Human Health and Disease. Int. J. Environ. Res. Public Health 2021, 18, 12836.
- Mattos, A.A.; Wiltgen, D.; Jotz, R.F.; Dornelles, C.; Fernandes, M.V.; Mattos, Z. Spontaneous bacterial peritonitis and extraperitoneal infections in patients with cirrhosis. Ann. Hepatol. 2020, 19, 451–457.
- Madrid, A.M.; Cumsille, F.; Defilippi, C. Altered small bowel motility in patients with liver cirrhosis depends on severity of liver disease. Dig. Dis. Sci. 1997, 42, 738–742.
- Huelin, P.; Piano, S.; Solà, E.; Stanco, M.; Solé, C.; Moreira, R.; Pose, E.; Fasolato, S.; Fabrellas, N.; de Prada, G.; et al. Validation of a staging system for acute kidney injury in patients with cirrhosis and association with acute-on-chronic liver failure. Clin. Gastroenterol. Hepatol. 2017, 15, 438–445.
- Wong, F.; Piano, S.; Singh, V.; Bartoletti, M.; Maiwall, R.; Alessandria, C.; Fernandez, J.; Soares, E.C.; Kim, D.J.; Kim, S.E.; et al. Clinical features and evolution of bacterial infection-related acute-on- chronic liver failure. J. Hepatol. 2020, 74, 330–339.
- Levesque, E.; Saliba, F.; Ichaï, P.; Samuel, D. Outcome of patients with cirrhosis requiring mechanical ventilation in ICU. J. Hepatol. 2014, 60, 570–578.
- Fernández, J.; Bert, F.; Nicolas-Chanoine, M.H. The challenges of multidrug-resistance in hepatology. J. Hepatol. 2016, 65, 1043–1054.
- Patel VC, Lee S, McPhail MJW, et al. Rifaximin-α reduces gut-derived inflammation and mucin degradation in cirrhosis and encephalopathy: RIFSYS randomised controlled trial. J Hepatol. 2022;76(2):332-342. doi:10.1016/j.jhep.2021.09.010
- Patel VC, Lee S, McPhail MJW, et al. Rifaximin-α reduces gut-derived inflammation and mucin degradation in cirrhosis and encephalopathy: RIFSYS randomised controlled trial.. J Hepatol. 2022, 76(2), 332-342, doi:10.1016/j.jhep.2021.09.010.
- Bajaj, J.S.; Reddy, K.R.; Tandon, P.; Garcia-Tsao, G.; Kamath, P.S.; O’Leary, J.G.; Wong, F.; Lai, J.; Vargas, H.; Thuluvath, P.J.; et al. Association of serum metabolites and gut microbiota at hospital admission with nosocomial infection development in patients with cirrhosis. Liver Transplant. 2022, 28, 1831–1840.
- Bernsmeier, C.; Van der Merwe, S.; Périanin, A. Innate immune cells in cirrhosis. J. Hepatol. 2020, 73, 186–201.
- Lebossé, F.; Gudd, C.; Tunc, E.; Singanayagam, A.; Nathwani, R.; Triantafyllou, E.; Pop, O.; Kumar, N.; Mukherjee, S.; Hou, T.Z.; et al. CD8+T cells from patients with cirrhosis display a phenotype that may contribute to cirrhosis-associated immune dysfunction. EBioMedicine 2019, 49, 258–268.
- Corrigall, D.; Harris, N.; Devshi, D.; Fairclough, S.; Wright, G.; Trebicka, J.; Alessandria, C.; Angeli, P.; Jalan, R.; Claria, J.; et al. First description of the immune checkpoint receptor landscape in decompensated cirrhosis & ACLF. J. Hepatol. 2019, 70, E175–E176.
- Bajaj, J.S.; Kamath, P.S.; Reddy, K.R. The evolving challenge of infections in cirrhosis. N. Engl. J. Med. 2021, 384, 2317–2330.
- Stengel, S.; Quickert, S.; Lutz, P.; Ibidapo-Obe, O.; Steube, A.; Köse-Vogel, N.; Yarbakht, M.; Reuken, P.A.; Busch, M.; Brandt, A.; et al. Peritoneal level of CD206 associates with mortality and an inflammatory macrophage phenotype in patients with decompensated cirrhosis and spontaneous bacterial peritonitis. Gastroenterology 2020, 158, 1745–1761.
- Porcheray, F.; Viaud, S.; Rimaniol, A.-C.; Léone, C.; Samah, B.; Dereuddre-Bosquet, N.; Dormont, D.; Gras, G. Macrophage activation switching: An asset for the resolution of inflammation. Clin. Exp. Immunol. 2005, 142, 481–489.
- Martinez-Pomares, L. The mannose receptor. J. Leukoc. Biol. 2012, 92, 1177–1186.
- Wang, C.; Ma, C.; Gong, L.; Guo, Y.; Fu, K.; Zhang, Y.; Zhou, H.; Li, Y. Macrophage polarization and its role in liver disease. Front. Immunol. 2021, 12, 5381.
- Bansal, R.; Van Baarlen, J.; Storm, G.; Prakash, J. The interplay of the Notch signaling in hepatic stellate cells and macrophages determines the fate of liver fibrogenesis. Sci. Rep. 2015, 5, 18272.
- Arvanitakis, K.; Koletsa, T.; Mitroulis, I.; Germanidis, G. Tumor-associated macrophages in hepatocellular carcinoma pathogenesis, prognosis and therapy. Cancers 2022, 14, 226.
- Li, R.; Zhou, R.; Wang, H.; Li, W.; Pan, M.; Yao, X.; Zhan, W.; Yang, S.; Xu, L.; Ding, Y.; et al. Gut microbiota-stimulated cathepsin K secretion mediates TLR4-dependent M2 macrophage polarization and promotes tumor metastasis in colorectal cancer. Cell Death Differ. 2019, 26, 2447–2463.
- Labonte, A.C.; Tosello-Trampont, A.C.; Hahn, Y.S. The role of macrophage polarization in infectious and inflammatory diseases. Mol. Cells 2014, 37, 275.
- Tsuchiya, K.; Suzuki, Y.; Yoshimura, K.; Yasui, H.; Karayama, M.; Hozumi, H.; Furuhashi, K.; Enomoto, N.; Fujisawa, T.; Nakamura, Y.; et al. Macrophage mannose receptor CD206 predicts prognosis in community-acquired pneumonia. Sci. Rep. 2019, 9, 18750.
- Van der Merwe, S.; Chokshi, S.; Bernsmeier, C.; Albillos, A. The multifactorial mechanisms of bacterial infection in decompensated cirrhosis. J. Hepatol. 2021, 75, S82–S100.
- Bernardi, M.; Angeli, P.; Claria, J.; Moreau, R.; Gines, P.; Jalan, R.; Caraceni, P.; Fernandez, J.; Gerbes, A.L.; O’Brien, A.J.; et al. Recent advances in clinical practice Albumin in decompensated cirrhosis: New concepts and perspectives. Gut 2020, 69, 1127–1138.
- Li, L.; Chen, L.; Lin, F.; Mu, J.; Wang, D.; Zhang, W.; Mi, L.; Qin, Y.; Zhou, Y. Study of the expression of inflammatory factors IL-4, IL-6, IL-10, and IL-17 in liver failure complicated by coagulation dysfunction and sepsis. J. Inflamm. Res. 2021, 14, 1447–1453.
- Piano, S.; Singh, V.; Caraceni, P.; Maiwall, R.; Alessandria, C.; Fernandez, J.; Soares, E.C.; Kim, D.J.; Kim, S.E.; Marino, M.; et al. Epidemiology and effects of bacterial infections in patients with cirrhosis worldwide. Gastroenterology 2019, 156, 1368–1380.
- Bajaj, J.S.; Garcia-Tsao, G.; Biggins, S.W.; Kamath, P.S.; Wong, F.; McGeorge, S.; Shaw, J.; Pearson, M.; Chew, M.; Fagan, A.; et al. Comparison of mortality risk in patients with cirrhosis and COVID-19 compared with patients with cirrhosis alone and COVID-19 alone: Multicentre matched cohort. Gut 2020, 70, 531–536.
- Fernández, J.; Prado, V.; Trebicka, J.; Amoros, A.; Gustot, T.; Wiest, R.; Deulofeu, C.; Garcia, E.; Acevedo, J.; Fuhrmann, V.; et al. Multidrug-resistant bacterial infections in patients with decompensated cirrhosis and with acute-on-chronic liver failure in Europe. J. Hepatol. 2019, 70, 398–411.
- Such, J.; Runyon, B.A. Spontaneous bacterial peritonitis. Clin. Infect. Dis. 1998, 27, 669–674.
- Casafont, F.; Sánchez, E.; Martin, L.; Aguero, J.; Romero, F.P. Influence of malnutrition on the prevalence of bacterial translocation and spontaneous bacterial peritonitis in experimental cirrhosis in rats. Hepatology 1997, 25, 1334–1337.
- FFriedrich, K.; Baumann, C.; Brune, M.; Wannhoff, A.; Rupp, C.; Scholl, S.G.; Antoni, C.; Dollinger, M.; Neumann-Haefelin, C.; Weiss, K.H.; et al. Association of serum zinc levels with liver function and survival in patients awaiting liver transplantation. Langenbeck’s Arch. Surg. 2015, 400, 805–811.
- Yousif, M.M.; Sadek, A.M.E.M.; Farrag, H.A.; Selim, F.O.; Hamed, E.F.; Salama, R.I. Associated vitamin D deficiency is a risk factor for the complication of HCV-related liver cirrhosis including hepatic encephalopathy and spontaneous bacterial peritonitis. Intern. Emerg. Med. 2019, 14, 753–761.
- Tergast, T.L.; Laser, H.; Gerbel, S.; Manns, M.P.; Cornberg, M.; Maasoumy, B. Association between type 2 diabetes mellitus, HbA1c and the risk for spontaneous bacterial peritonitis in patients with decompensated liver cirrhosis and ascites. Clin. Transl. Gastroenterol. 2018, 9, e189.
- Nicoletti, A.; Ponziani, F.R.; Biolato, M.; Valenza, V.; Marrone, G.; Sganga, G.; Gasbarrini, A.; Miele, L.; Grieco, A. Intestinal permeability in the pathogenesis of liver damage: From non-alcoholic fatty liver disease to liver transplantation. World J. Gastroenterol. 2019, 25, 4814–4834.
- Paradis, T.; Bègue, H.; Basmaciyan, L.; Dalle, F.; Bon, F. Tight Junctions as a Key for Pathogens Invasion in Intestinal Epithelial Cells. Int. J. Mol. Sci. 2021, 22, 2506.
- Aguirre Valadez, J.M.; Rivera-Espinosa, L.; Méndez-Guerrero, O.; Chávez-Pacheco, J.L.; García Juárez, I.; Torre, A. Intestinal permeability in a patient with liver cirrhosis. Ther. Clin. Risk Manag. 2016, 12, 1729–1748.
- Wiest, R.; Lawson, M.; Geuking, M. Pathological bacterial translocation in liver cirrhosis. J. Hepatol. 2014, 60, 197–209.
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411.
- Qin, N.; Yang, F.; Li, A.; Prifti, E.; Chen, Y.; Shao, L.; Guo, J.; Le Chatelier, E.; Yao, J.; Wu, L.; et al. Alterations of the human gut microbiome in liver cirrhosis. Nature 2014, 513, 59–64.
- Ponziani, F.R.; Nicoletti, A.; Gasbarrini, A.; Pompili, M. Diagnostic and therapeutic potential of the gut microbiota in patients with early hepatocellular carcinoma. Ther. Adv. Med. Oncol. 2019, 11, 1758835919848184.
- Solé, C.; Guilly, S.; Da Silva, K.; Llopis, M.; Le-Chatelier, E.; Huelin, P.; Ginès, P. Alterations in gut microbiome in cirrhosis as assessed by quantitative metagenomics: Relationship with acute-on-chronic liver failure and prognosis. Gastroenterology 2021, 160, 206–218.
- Bajaj, J.S.; Heuman, D.M.; Hylemon, P.B.; Sanyal, A.J.; White, M.B.; Monteith, P.; Noble, N.A.; Unser, A.B.; Daita, K.; Fisher, A.R.; et al. Altered profile of human gut microbiome is associated with cirrhosis and its complications. J. Hepatol. 2014, 60, 940–947.
- Bajaj, J.S.; Hylemon, P.B.; Ridlon, J.M.; Heuman, D.M.; Daita, K.; White, M.B.; Monteith, P.; Noble, N.A.; Sikaroodi, M.; Gillevet, P.M. Colonic mucosal microbiome differs from stool microbiome in cirrhosis and hepatic encephalopathy and is linked to cognition and inflammation. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 303, 675–685.
- Feng, Y.; Chen, C.L.; Chen, T.H.; Liang, Y.H.; Chen, H.L.; Lin, C.Y.; Chiu, C.H. Application of next-generation sequencing to study ascitic microbiome in cirrhotic patients with or without spontaneous bacterial peritonitis. J. Microbiol. 2015, 48, 504–509.
- Guarner, C.; Soriano, G.; Tomas, A.; Bulbena, O.; Novella, M.T.; Balanzo, J.; Vilardell, F.; Mourelle, M.; Moncada, S. Increased serum nitrite and nitrate levels in patients with cirrhosis: Relationship to endotoxemia. Hepatology 1993, 18, 1139–1143.
- Bajaj, J.S.; Rodriguez, M.P.; Fagan, A.; McGeorge, S.; Sterling, R.K.; Lee, H.; Gillevet, P.M. Impact of Bacterial Infections and SBP Prophylaxis on Phage-Bacterial Dynamics in Cirrhosis. Hepatology 2022, 76, 1723–1734.
- Zhou, Z.; Lv, H.; Lv, J.; Shi, Y.; Huang, H.; Chen, L.; Shi, D. Alterations of gut microbiota in cirrhotic patients with spontaneous bacterial peritonitis: A distinctive diagnostic feature. Front. Cell Infect. Microbiol. 2022, 12, 999418.
- Mücke, M.M.; Rüschenbaum, S.; Mayer, A.; Mücke, V.T.; Schwarzkopf, K.M.; Zeuzem, S.; Lange, C.M. Stool and sputum microbiome during quinolone prophylaxis of spontaneous bacterial peritonitis: An exploratory study. Gut Pathog. 2020, 12, 51.
- Ponziani, F.R.; Gerardi, V.; Pecere, S.; D’Aversa, F.; Lopetuso, L.; Zocco, M.A.; Pompili, M.; Gasbarrini, A. Effect of rifaximin on gut microbiota composition in advanced liver disease and its complications. World J. Gastroenterol. 2015, 21, 12322–12333.
- Caraceni, P.; Vargas, V.; Solà, E.; Alessandria, C.; de Wit, K.; Trebicka, J.; Watson, H. The use of rifaximin in patients with cirrhosis. Hepatology 2021, 74, 1660–1673.
- Patel VC, Lee S, McPhail MJW, et al. Rifaximin-α reduces gut-derived inflammation and mucin degradation in cirrhosis and encephalopathy: RIFSYS randomised controlled trial. J Hepatol. 2022;76(2):332-342. doi:10.1016/j.jhep.2021.09.010
- Elfert, A.; Abo Ali, L.; Soliman, S.; Ibrahim, S.; Abd-Elsalam, S. Randomized-controlled trial of rifaximin versus norfloxacin for secondary prophylaxis of spontaneous bacterial peritonitis. Eur. J. Gastroenterol. Hepatol. 2016, 28, 1450–1454.
- Lo, R.S.; Austin, A.S.; Freeman, J.G. Is there a role for probiotics in liver disease? Sci. World J. 2014, 2014, 874768.
- Wiest, R.; Chen, F.; Cadelina, G.; Groszmann, R.J.; Garcia-Tsao, G. Effect of Lactobacillus-fermented diets on bacterial translocation and intestinal flora in experimental prehepatic portal hypertension. Dig. Dis. Sci. 2003, 48, 1136–1141.
- Pande, C.; Kumar, A.; Sarin, S.K. Addition of probiotics to norfloxacin does not improve efficacy in the prevention of spontaneous bacterial peritonitis: A double-blind placebo-controlled randomized-controlled trial. Eur. J. Gastroenterol. Hepatol. 2012, 24, 831–839.
- Markowiak, P.; Śliżewska, K. Effects of Probiotics, Prebiotics, and Synbiotics on Human Health. Nutrients 2017, 9, 1021.
- Potruch, A.; Schwartz, A.; Ilan, Y. The role of bacterial translocation in sepsis: A new target for therapy. Ther. Adv. Gastroenterol. 2022, 15, 17562848221094214.
- Rose, E.C.; Odle, J.; Blikslager, A.T.; Ziegler, A.L. Probiotics, prebiotics and epithelial tight junctions: A promising approach to modulate intestinal barrier function. Int. J. Mol. Sci. 2021, 22, 6729.
- Jiang, H.; Peng, Y.; Zhang, W.; Chen, Y.; Jiang, Q.; Zhou, Y. Gut microbiome-targeted therapies in liver cirrhosis: A protocol for systematic review and meta-analysis. Syst. Rev. 2022, 11, 181.
- Zacharias, H.D.; Zacharias, A.P.; Gluud, L.L.; Morgan, M.Y. Pharmacotherapies that specifically target ammonia for the prevention and treatment of hepatic encephalopathy in adults with cirrhosis. Cochrane Database Syst. Rev. 2019, 6, CD012334.
- Macnaughtan, J.; Ranchal, I.; Soeda, J.; Sawhney, R.; Oben, J.; Davies, N.; Mookerjee, R.; Marchesi, J.; Cox, J.; Jalan, R. Oral therapy with non-absorbable carbons of controlled porosity (Yaq-001) selectively modulates stool microbiome and its function and this is associated with restoration of immune function and inflammasome activation. J. Hepatol. 2015, 62, S240.
- Ubeda, M.; Lario, M.; Munoz, L.; Borrero, M.J.; Rodriguez-Serrano, M.; Sanchez-Diaz, A.M.; Del Campo, R.; Lledó, L.; Pastor, Ó.; García-Bermejo, L.; et al. Obeticholic acid reduces bacterial translocation and inhibits intestinal inflammation in cirrhotic rats. J. Hepatol. 2016, 64, 1049–1057.
- Tsuji, Y.; Kaji, K.; Kitade, M.; Kaya, D.; Kitagawa, K.; Ozutsumi, T.; Fujinaga, Y.; Takaya, H.; Kawaratani, H.; Namisaki, T.; et al. Bile acid sequestrant, sevelamer ameliorates hepatic fibrosis with reduced overload of endogenous lipopolysaccharide in experimental nonalcoholic steatohepatitis. Microorganisms 2020, 8, 925.
- Sandhu, B.S.; Gupta, R.; Sharma, J.; Singh, J.; Murthy, N.S.; Sarin, S.K. Norfloxacin and cisapride combination decreases the incidence of spontaneous bacterial peritonitis in cirrhotic ascites. J. Gastroenterol. Hepatol. 2005, 20, 599–605.
- Pérez-Paramo, M.; Muñoz, J.; Albillos, A.; Freile, I.; Portero, F.; Santos, M.; Ortiz-Berrocal, J. Effect of propranolol on factors promoting bacterial translocation in cirrhotic rats with ascites. Hepatology 2000, 31, 43–48.
- Tranah, T.H.; Edwards, L.A.; Schnabl, B.; Shawcross, D.L. Targeting the gut-liver-immune axis to treat cirrhosis. Gut 2021, 70, 982–994.