Pulmonary vascular remodeling is the critical structural alteration and pathological feature in pulmonary hypertension (PH) and involves changes in the intima, media and adventitia. Pulmonary vascular remodeling consists of the proliferation and phenotypic transformation of pulmonary artery endothelial cells (PAECs) and pulmonary artery smooth muscle cells (PASMCs) of the middle membranous pulmonary artery, as well as complex interactions involving external layer pulmonary artery fibroblasts (PAFs) and extracellular matrix (ECM). Inflammatory mechanisms,apoptosis and other factors in the vascular wall are influenced by different mechanisms that likely act in concert to drive disease progression. This article reviews thesse pathological changes and highlights some pathogenetic mechanisms involved in the remodeling process are described.
- pulmonary hypertension
- pulmonary vascular remodeling
1. Introduction
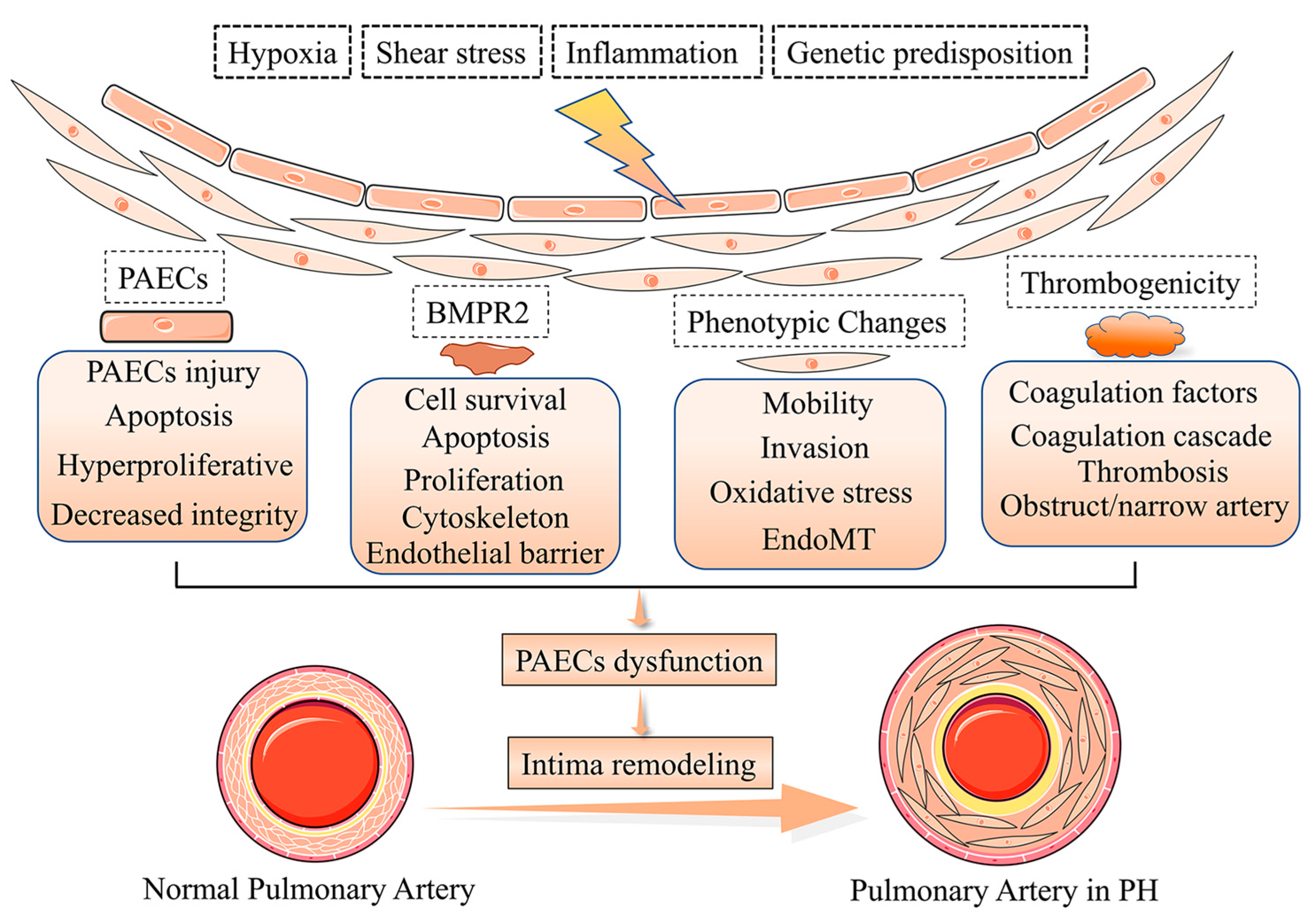
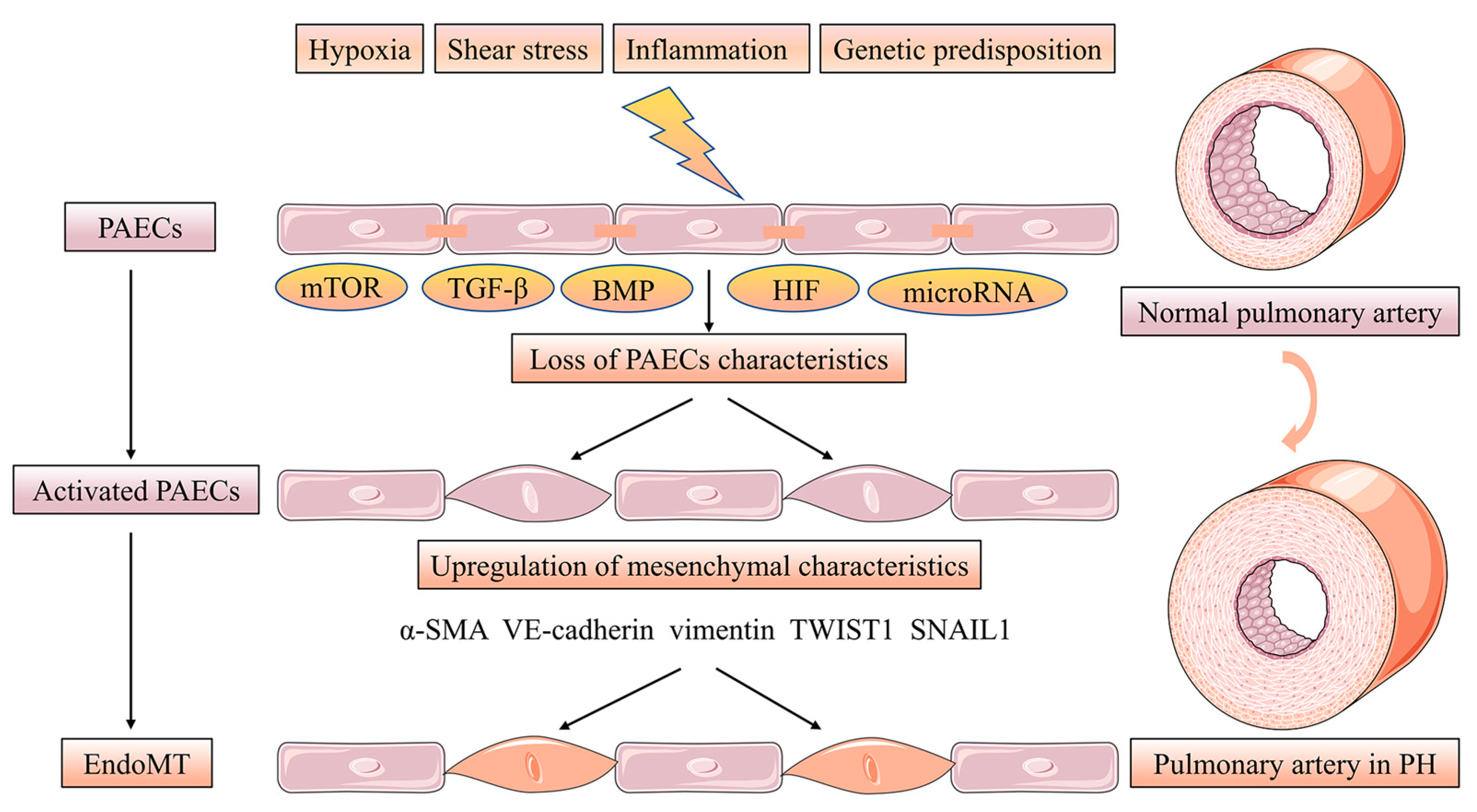
2. Phenotypes of Pulmonary Artery Endothelial Cells Dysfunction in Intima Remodeling
3. Pulmonary Artery Endothelial Cells Survival and Proliferation in Intima Remodeling
4. Pulmonary Artery Endothelial Cells Activation and Thrombogenicity in Intima Remodeling
5. Pulmonary Artery Endothelial Cells Metabolism and Epigenetics in Intima Remodeling
References
- Tuder, R.M. Pulmonary vascular remodeling in pulmonary hypertension. Cell Tissue Res. 2017, 367, 643–649.
- Stacher, E.; Graham, B.B.; Hunt, J.M.; Gandjeva, A.; Groshong, S.D.; McLaughlin, V.V.; Jessup, M.; Grizzle, W.E.; Aldred, M.A.; Cool, C.D.; et al. Modern age pathology of pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2012, 186, 261–272.
- Evans, C.E.; Cober, N.D.; Dai, Z.; Stewart, D.J. Endothelial cells in the pathogenesis of pulmonary arterial hypertension. Eur. Respir. J. 2021, 58, 2003957.
- Nie, X.; Shen, C.; Tan, J.; Wu, Z.; Wang, W.; Chen, Y.; Dai, Y.; Yang, X.; Ye, S.; Chen, J.; et al. Periostin: A Potential Therapeutic Target For Pulmonary Hypertension? Circ. Res. 2020, 127, 1138–1152.
- Dummer, A.; Rol, N.; Szulcek, R.; Kurakula, K.; Pan, X.; Visser, B.I.; Bogaard, H.J.; DeRuiter, M.C.; Goumans, M.J.; Hierck, B.P. Endothelial dysfunction in pulmonary arterial hypertension: Loss of cilia length regulation upon cytokine stimulation. Pulm. Circ. 2018, 8, 2045894018764629.
- Gorelova, A.; Berman, M.; Al Ghouleh, I. Endothelial-to-Mesenchymal Transition in Pulmonary Arterial Hypertension. Antioxid. Redox Signal. 2021, 34, 891–914.
- Rodor, J.; Chen, S.H.; Scanlon, J.P.; Monteiro, J.P. Single-cell RNA sequencing profiling of mouse endothelial cells in response to pulmonary arterial hypertension. Cardiovasc. Res. 2022, 118, 2519–2534.
- Ranchoux, B.; Harvey, L.D.; Ayon, R.J.; Babicheva, A.; Bonnet, S.; Chan, S.Y.; Yuan, J.X.; Perez, V.J. Endothelial dysfunction in pulmonary arterial hypertension: An evolving landscape (2017 Grover Conference Series). Pulm. Circ. 2018, 8, 2045893217752912.
- Bochenek, M.L.; Rosinus, N.S.; Lankeit, M.; Hobohm, L.; Bremmer, F.; Schütz, E.; Klok, F.A.; Horke, S.; Wiedenroth, C.B.; Münzel, T.; et al. From thrombosis to fibrosis in chronic thromboembolic pulmonary hypertension. Thromb. Haemost. 2017, 117, 769–783.
- Chabert, C.; Khochbin, S.; Rousseaux, S.; Veyrenc, S. Inhibition of BET Proteins Reduces Right Ventricle Hypertrophy and Pulmonary Hypertension Resulting from Combined Hypoxia and Pulmonary Inflammation. Int. J. Mol. Sci. 2018, 19, 2224.
- Hautefort, A.; Mendes-Ferreira, P.; Sabourin, J.; Manaud, G.; Bertero, T.; Rucker-Martin, C.; Riou, M.; Adão, R.; Manoury, B.; Lambert, M.; et al. Bmpr2 Mutant Rats Develop Pulmonary and Cardiac Characteristics of Pulmonary Arterial Hypertension. Circulation 2019, 139, 932–948.
- Morrell, N.W.; Aldred, M.A.; Chung, W.K.; Elliott, C.G.; Nichols, W.C.; Soubrier, F. Genetics and genomics of pulmonary arterial hypertension. Eur. Respir. J. 2019, 53, 1801899.
- Ruffenach, G.; O’Connor, E.; Vaillancourt, M.; Hong, J.; Cao, N.; Sarji, S.; Moazeni, S.; Papesh, J.; Grijalva, V.; Cunningham, C.M.; et al. Oral 15-Hydroxyeicosatetraenoic Acid Induces Pulmonary Hypertension in Mice by Triggering T Cell-Dependent Endothelial Cell Apoptosis. Hypertension 2020, 76, 985–996.
- Sakao, S.; Tatsumi, K.; Voelkel, N.F. Endothelial cells and pulmonary arterial hypertension: Apoptosis, proliferation, interaction and transdifferentiation. Respir. Res. 2009, 10, 95.
- Kim, C.; Seedorf, G.J.; Abman, S.H.; Shepherd, D.P. Heterogeneous response of endothelial cells to insulin-like growth factor 1 treatment is explained by spatially clustered sub-populations. Biol. Open 2019, 8, bio045906.
- Sánchez-Duffhues, G.; García de Vinuesa, A.; Ten Dijke, P. Endothelial-to-mesenchymal transition in cardiovascular diseases: Developmental signaling pathways gone awry. Dev. Dyn. Off. Publ. Am. Assoc. Anat. 2018, 247, 492–508.
- Mammoto, T.; Muyleart, M.; Konduri, G.G.; Mammoto, A. Twist1 in Hypoxia-induced Pulmonary Hypertension through Transforming Growth Factor-β-Smad Signaling. Am. J. Respir. Cell Mol. Biol. 2018, 58, 194–207.
- Ursoli Ferreira, F.; Eduardo Botelho Souza, L.; Hassibe Thomé, C.; Tomazini Pinto, M.; Origassa, C.; Salustiano, S.; Marcel Faça, V.; Olsen Câmara, N.; Kashima, S.; Tadeu Covas, D. Endothelial Cells Tissue-Specific Origins Affects Their Responsiveness to TGF-β2 during Endothelial-to-Mesenchymal Transition. Int. J. Mol. Sci. 2019, 20, 458.
- Zhang, H.; Liu, Y.; Yan, L.; Du, W.; Zhang, X.; Zhang, M.; Chen, H.; Zhang, Y.; Zhou, J.; Sun, H.; et al. Bone morphogenetic protein-7 inhibits endothelial-mesenchymal transition in pulmonary artery endothelial cell under hypoxia. J. Cell. Physiol. 2018, 233, 4077–4090.
- Hiepen, C.; Jatzlau, J.; Hildebrandt, S.; Kampfrath, B.; Goktas, M. BMPR2 acts as a gatekeeper to protect endothelial cells from increased TGFβ responses and altered cell mechanics. PLoS Biol. 2019, 17, e3000557.
- Rol, N.; Kurakula, K.B.; Happé, C.; Bogaard, H.J.; Goumans, M.J. TGF-β and BMPR2 Signaling in PAH: Two Black Sheep in One Family. Int. J. Mol. Sci. 2018, 19, 2585.
- Dai, Z.; Zhu, M.M.; Peng, Y.; Machireddy, N.; Evans, C.E.; Machado, R.; Zhang, X.; Zhao, Y.Y. Therapeutic Targeting of Vascular Remodeling and Right Heart Failure in Pulmonary Arterial Hypertension with a HIF-2α Inhibitor. Am. J. Respir. Crit. Care Med. 2018, 198, 1423–1434.
- Liu, T.; Zou, X.Z.; Huang, N.; Ge, X.Y.; Yao, M.Z.; Liu, H.; Zhang, Z.; Hu, C.P. miR-27a promotes endothelial-mesenchymal transition in hypoxia-induced pulmonary arterial hypertension by suppressing BMP signaling. Life Sci. 2019, 227, 64–73.
- Zhang, H.; Wang, D.; Li, M.; Plecitá-Hlavatá, L.; D’Alessandro, A.; Tauber, J.; Riddle, S.; Kumar, S.; Flockton, A.; McKeon, B.A.; et al. Metabolic and Proliferative State of Vascular Adventitial Fibroblasts in Pulmonary Hypertension Is Regulated Through a MicroRNA-124/PTBP1 (Polypyrimidine Tract Binding Protein 1)/Pyruvate Kinase Muscle Axis. Circulation 2017, 136, 2468–2485.
- Zhao, H.; Wang, Y.; Zhang, X.; Guo, Y.; Wang, X. miR-181b-5p inhibits endothelial-mesenchymal transition in monocrotaline-induced pulmonary arterial hypertension by targeting endocan and TGFBR1. Toxicol. Appl. Pharmacol. 2020, 386, 114827.
- Happé, C.; Kurakula, K.; Sun, X.Q.; da Silva Goncalves Bos, D. The BMP Receptor 2 in Pulmonary Arterial Hypertension: When and Where the Animal Model Matches the Patient. Cells 2020, 9, 1422.
- Bisserier, M.; Mathiyalagan, P.; Zhang, S.; Elmastour, F.; Dorfmüller, P.; Humbert, M.; David, G.; Tarzami, S.; Weber, T.; Perros, F.; et al. Regulation of the Methylation and Expression Levels of the BMPR2 Gene by SIN3a as a Novel Therapeutic Mechanism in Pulmonary Arterial Hypertension. Circulation 2021, 144, 52–73.
- Hodgson, J.; Swietlik, E.M.; Salmon, R.M.; Hadinnapola, C.; Nikolic, I.; Wharton, J.; Guo, J.; Liley, J.; Haimel, M.; Bleda, M.; et al. Characterization of GDF2 Mutations and Levels of BMP9 and BMP10 in Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2020, 201, 575–585.
- Rol, N.; de Raaf, M.A.; Sun, X.Q.; Kuiper, V.P.; da Silva Gonçalves Bos, D.; Happé, C.; Kurakula, K.; Dickhoff, C.; Thuillet, R.; Tu, L.; et al. Nintedanib improves cardiac fibrosis but leaves pulmonary vascular remodelling unaltered in experimental pulmonary hypertension. Cardiovasc. Res. 2019, 115, 432–439.
- Li, C.G.; Mahon, C.; Sweeney, N.M.; Verschueren, E.; Kantamani, V.; Li, D.; Hennigs, J.K.; Marciano, D.P.; Diebold, I.; Abu-Halawa, O.; et al. PPARγ Interaction with UBR5/ATMIN Promotes DNA Repair to Maintain Endothelial Homeostasis. Cell Rep. 2019, 26, 1333–1343.e1337.
- Tang, H.; Babicheva, A.; McDermott, K.M.; Gu, Y.; Ayon, R.J.; Song, S.; Wang, Z.; Gupta, A.; Zhou, T.; Sun, X.; et al. Endothelial HIF-2α contributes to severe pulmonary hypertension due to endothelial-to-mesenchymal transition. American journal of physiology. Lung Cell. Mol. Physiol. 2018, 314, L256–L275.
- Oliveira, S.D.S.; Chen, J.; Castellon, M.; Mao, M.; Raj, J.U.; Comhair, S.; Erzurum, S.; Silva, C.L.M.; Machado, R.F.; Bonini, M.G.; et al. Injury-Induced Shedding of Extracellular Vesicles Depletes Endothelial Cells of Cav-1 (Caveolin-1) and Enables TGF-β (Transforming Growth Factor-β)-Dependent Pulmonary Arterial Hypertension. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1191–1202.
- Yu, X.; Chen, X.; Zheng, X.D.; Zhang, J.; Zhao, X.; Liu, Y.; Zhang, H.; Zhang, L.; Yu, H.; Zhang, M.; et al. Growth Differentiation Factor 11 Promotes Abnormal Proliferation and Angiogenesis of Pulmonary Artery Endothelial Cells. Hypertension 2018, 71, 729–741.
- Kurakula, K.; Sun, X.Q.; Happé, C.; da Silva Goncalves Bos, D.; Szulcek, R.; Schalij, I.; Wiesmeijer, K.C.; Lodder, K.; Tu, L.; Guignabert, C. Prevention of progression of pulmonary hypertension by the Nur77 agonist 6-mercaptopurine: Role of BMP signalling. Eur. Respir. J. 2019, 54, 1802400.
- Wang, E.L.; Jia, M.M.; Luo, F.M.; Li, T.; Peng, J.J.; Luo, X.J.; Song, F.L.; Yang, J.F.; Peng, J.; Liu, B. Coordination between NADPH oxidase and vascular peroxidase 1 promotes dysfunctions of endothelial progenitor cells in hypoxia-induced pulmonary hypertensive rats. Eur. J. Pharmacol. 2019, 857, 172459.
- Goyanes, A.M.; Moldobaeva, A.; Marimoutou, M.; Varela, L.C.; Wang, L.; Johnston, L.F.; Aladdin, M.M.; Peloquin, G.L.; Kim, B.S.; Damarla, M.; et al. Functional Impact of Human Genetic Variants of COL18A1/Endostatin on Pulmonary Endothelium. Am. J. Respir. Cell Mol. Biol. 2020, 62, 524–534.
- Park, C.S.; Kim, S.H.; Yang, H.Y.; Kim, J.H.; Schermuly, R.T.; Cho, Y.S.; Kang, H.; Park, J.H.; Lee, E. Sox17 Deficiency Promotes Pulmonary Arterial Hypertension via HGF/c-Met Signaling. Circ. Res. 2022, 131, 792–806.
- Kondababu, K.; Smolders, V.F.E.D.; Olga, T.; Wouter, J.J.; Quax, P.H.A.; MarieJosé, G. Endothelial Dysfunction in Pulmonary Hypertension: Cause or Consequence? Biomedicines 2021, 9, 57.
- Sakamaki, F.; Kyotani, S.; Nagaya, N.; Sato, N.; Oya, H.; Satoh, T.; Nakanishi, N. Increased plasma P-selectin and decreased thrombomodulin in pulmonary arterial hypertension were improved by continuous prostacyclin therapy. Circulation 2000, 102, 2720–2725.
- Maeda, N.Y.; Clavé, M.M.; Bydlowski, S.P.; Lopes, A.A. Decreased circulating thrombomodulin is improved by tadalafil therapy in hypoxemic patients with advanced pulmonary arterial hypertension. Thromb. Res. 2016, 146, 15–19.
- Pan, Y.Y.; Yang, J.X.; Mao, W.; Wang, X.X. RNA-binding protein SFPQ cooperates with HDAC1 to suppress CD40 transcription in pulmonary adventitial fibroblasts. Cell Biol. Int. 2019, 44, 166–176.
- Kovacs, L.; Cao, Y.; Han, W.; Meadows, L.; Kovacs-Kasa, A.; Kondrikov, D.; Verin, A.D.; Barman, S.A.; Dong, Z.; Huo, Y.; et al. PFKFB3 in Smooth Muscle Promotes Vascular Remodeling in Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2019, 200, 617–627.
- Sun, X.; Kumar, S.; Sharma, S.; Aggarwal, S.; Lu, Q.; Gross, C.; Rafikova, O.; Lee, S.G.; Dasarathy, S.; Hou, Y.; et al. Endothelin-1 induces a glycolytic switch in pulmonary arterial endothelial cells via the mitochondrial translocation of endothelial nitric oxide synthase. Am. J. Respir. Cell Mol. Biol. 2014, 50, 1084–1095.
- Yu, Q.; Tai, Y.Y.; Tang, Y.; Zhao, J.; Negi, V.; Culley, M.K.; Pilli, J.; Sun, W.; Brugger, K.; Mayr, J.; et al. BOLA (BolA Family Member 3) Deficiency Controls Endothelial Metabolism and Glycine Homeostasis in Pulmonary Hypertension. Circulation 2019, 139, 2238–2255.
- Bertero, T.; Oldham, W.M.; Cottrill, K.A.; Pisano, S.; Vanderpool, R.R.; Yu, Q.; Zhao, J.; Tai, Y.; Tang, Y.; Zhang, Y.Y.; et al. Vascular stiffness mechanoactivates YAP/TAZ-dependent glutaminolysis to drive pulmonary hypertension. J. Clin. Investig. 2016, 126, 3313–3335.
- Thoré, P.; Girerd, B.; Jaïs, X.; Savale, L.; Ghigna, M.R.; Eyries, M.; Levy, M.; Ovaert, C.; Servettaz, A. Phenotype and outcome of pulmonary arterial hypertension patients carrying a TBX4 mutation. Eur. Respir. J. 2020, 55, 1902340.