Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Rita Xu and Version 1 by Clément Lemaitre.
Atropisomers are fascinating objects of study by themselves for chemists but also find applications in various sub-fields of applied chemistry. Among these strategies, oxidative aromatization with central-to-axial conversion of chirality has gained increasing popularity. It consists of the oxidation of a cyclic non-aromatic precursors into the corresponding aromatic atropisomers.
- aromatic compounds
- atropisomers
- conversion of chirality
1. Introduction
1.1. Interest of Atropisomers and Synthetic Methods to Prepare Them
Described for the first time 100 hundred years ago [1], atropisomers are conformers that gained enough stability because of the hindered rotation around a single bond. They are, thus, considered configurational stereoisomers [2], in which the bond with hindered rotation becomes a stereogenic axis. A half time life higher than 103 s is usually set as the limit to claim their existence, corresponding to a barrier to rotation of 93 kJ·mol−1 at 300 K [2,3][2][3]. However, to be synthesized, stored, and used at ambient or biological temperature, higher barriers to rotation of ≥105 kJ·mol−1 are generally required. The most frequently encountered atropisomers are biaryl compounds bearing ortho substituents to prevent the rotation around the biaryl bond. However, non-biaryl based atropisomers also exist [4,5,6][4][5][6].
In view of their unique properties, atropisomers have received much attention from researchers in all sub-fields of chemistry (Figure 1) [7,8,9][7][8][9]. They are present in natural products [10[10][11],11], such as gossypol (1) [12], and bioactive compounds [13,14,15,16,17][13][14][15][16][17], such as HIV integrase inhibitor (2) [18] and kinase inhibitor (3) [19], but they also find applications as metal ligands, such as BINAP (4) [20], organocatalysts, such as chiral phosphoric acid (5) [21], in polymer science [22[22][23],23], etc.
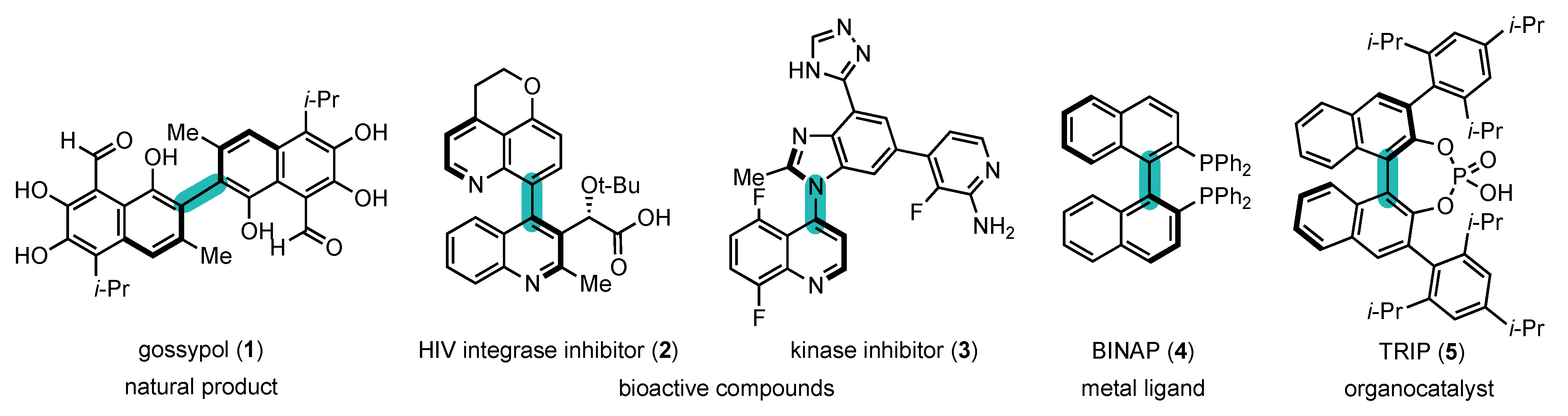
Figure 1. Atropisomers: examples and their fields of application.
Considering the importance of atropisomers and the fact that the two enantiomers are likely to have different behaviors when placed in a chiral environment (as is the case for biological applications), methods to obtain them in enantioenriched form have been actively pursued. While resolution of the racemate is often used, the development of enantioselective synthetic methods can be highly appealing since (i) they obviate the need to discard the unwanted enantiomer or recycle it by enantiomerization; and (ii) they stimulate the creativity of organic chemists.
Enantioenriched atropisomers can be synthesized by a variety of methods, which can be classified into six main categories (Scheme 1): [24,25,26,27,28,29,30][24][25][26][27][28][29][30]
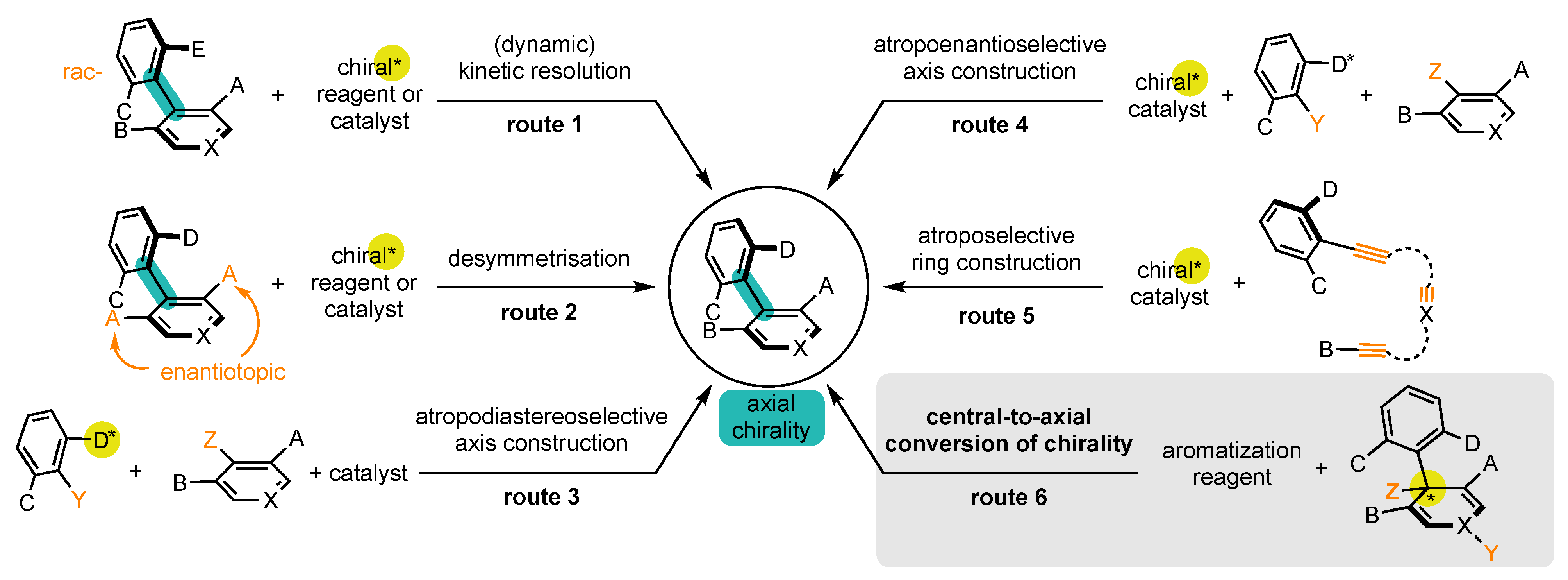
Scheme 1. Synthetic strategies to prepare atropisomers (presented on biaryl atropisomers for better readability).
- ]
- .
- Route 6:
It should be noted that routes 1 and 2 start from substrates where the bond of the stereogenic axis and the two aromatic rings already preexist. On the contrary, substrates where the bond does not exist are involved in routes 3 and 4, whereas routes 5 and 6 involve substrates where at least one aromatic ring has to be constructed.
1.2. Defining Conversion of Chirality
WResearchers wish to define as conversions of chirality [51][50] all synthetic operations where a stereogenic element is forged in a molecule concomitantly with the destruction of a stereogenic element of a different type (Scheme 2a). The selection of this name requires an in-depth explanation:
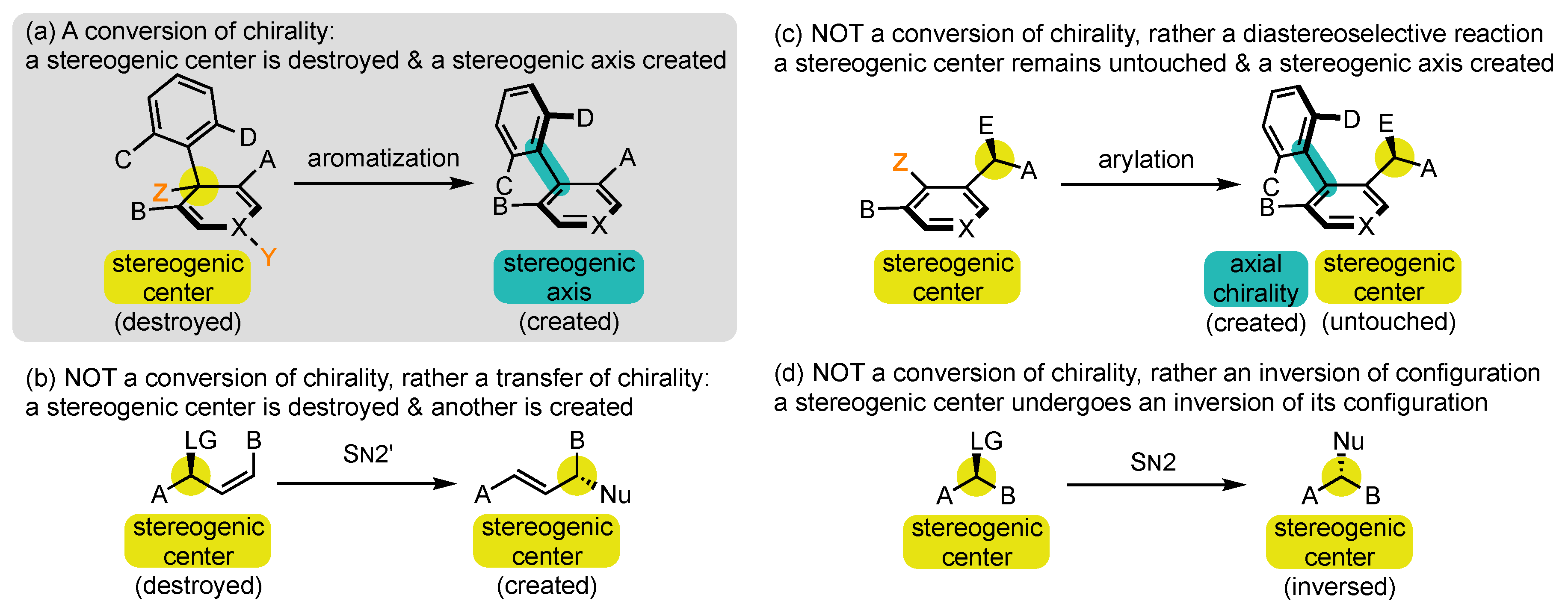
Scheme 2. What is and is NOT a conversion of chirality?
-
At first, chirality is used in a quite improper sense, as it is normally applied to a molecule or an object in its entirety, while the process discussed here falls within local stereochemistry discussions [52][51]. Conversion of stereogenicity should, in fact, be recommended, but it seems completely absent from the literature and perhaps more difficult to comprehend for those who are not specialists of this field.
-
Conversions of chirality are also referred to in the literature by several other denominations, which all appear to uresearchers as less suited. Transfer has long been the most frequently used term [53,54,55][52][53][54], but it brings the idea of relocation rather than transformation. For uresearchers, it should be restricted to reactions where the stereogenic element that is destroyed and the one that is created are of the same nature, such as in SN2′ reactions with allylic leaving groups (Scheme 2b)Scheme 2b) [55]. [56].Exchange Exchange [49,57][49][56] and interconversion and interconversion[47] can also be encountered, but they evoke the idea of reversibility, which is generally not the case in those [47] can also be encountered, but they evoke the idea of reversibility, which is generally not the case in those transformations.transformations.
Having defined the conversion of chirality, wresearchers now wish to point out which type of phenomenon should NOT be called so. Indeed, all the terms presented above are often misused to describe phenomena that are fundamentally different from conversion of chirality. Conversions/transfers/interconversions/exchanges of chirality are by nature potentially stereospecific processes and should be clearly distinguished from diastereoselective reactions. WResearchers can, for example, cite those where the configuration of a stereogenic axis is controlled thanks to the presence of one or several stereogenic centers in the surroundings, which remain completely untouched during the transformation (Scheme 2c) [58,59,60,61,62][57][58][59][60][61]. Rather, they belong to the strategy exposed in Route 3 above. Sometimes these terms are even used for reactions that involve only an inversion of configuration, such as in a SN2 reaction (Scheme 2d) [63][62].
Being a phenomenon of enantiospecific nature, the efficiency of a conversion of chirality should be evaluated by comparing the relative enantioenrichment of the product and the substrate. In order to normalize this evaluation, Bressy, Bugaut, and Rodriguez introduced the notion of conversion percentage (cp) [64][63], which can be calculated as such and will be used throughout this review:
2. Historical Background
2.1. A surprisingly Early First Example!
From an historical point of view, it is quite amusing to realize that the first central-to-axial conversion of chirality was achieved in 1905 by Freund, even before the description of axial chirality, during reactivity studies on the natural product thebaine (6) (Scheme 3) [65][64]. Of course, it could not be identified at that moment, and it took almost half a century before the real explanation of what happened could be given by Robinson and Berson [66,67,68][65][66][67]. Treatment of 6 with phenylmagnesium bromide resulted in a complex rearrangement with aromatization to deliver biaryl atropisomer 9 as a mixture of two diastereomers. The reaction proceeds through the opening of the oxa-bridge with rearrangement of the carbon skeleton to afford 7, which can aromatize to 8. At this stage, the three initially present stereogenic centers have been destroyed with the concomitant installation of the stereogenic axis. The reaction terminates with trapping of the iminium ion by the Grignard reagent, giving rise to the formation of 9 as two diastereomers, which complicated the understanding of the transformation.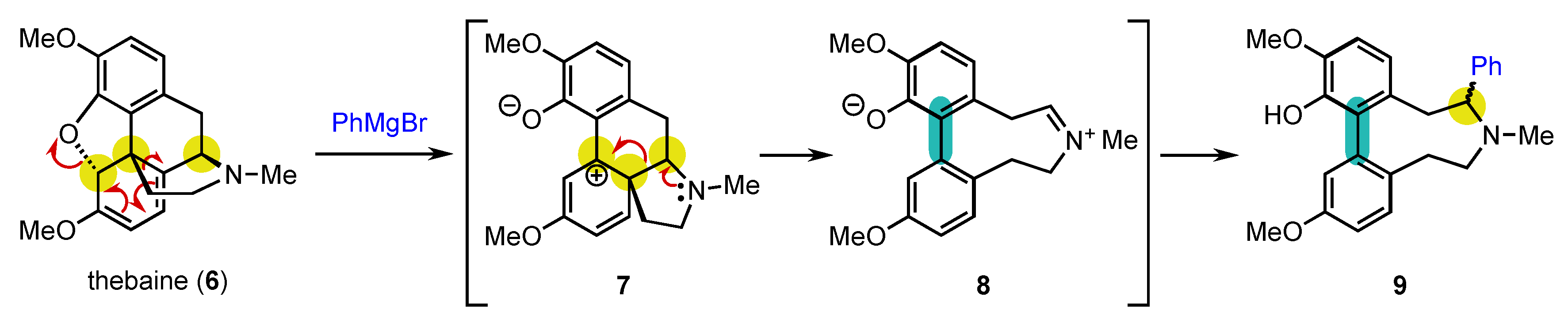
Scheme 3. Rearrangement of thebaine: the first example of central-to-axial conversion of chirality during aromatization.
2.2. Formulating Research Hypotheses to Achieve Conversion of Chirality
With the understanding of this phenomenon, Berson formulated three fundamental conditions that must be satisfied to allow the synthesis of atropisomers with central-to-axial conversion of chirality [51][50]:-
Centrally chiral non-aromatic precursors should be accessible in enantioenriched form;
-
Steric hindrance around the stereogenic axis should be sufficient to ensure its configurational stability;
-
Aromatization should occur with a preservation of the enantiopurity.
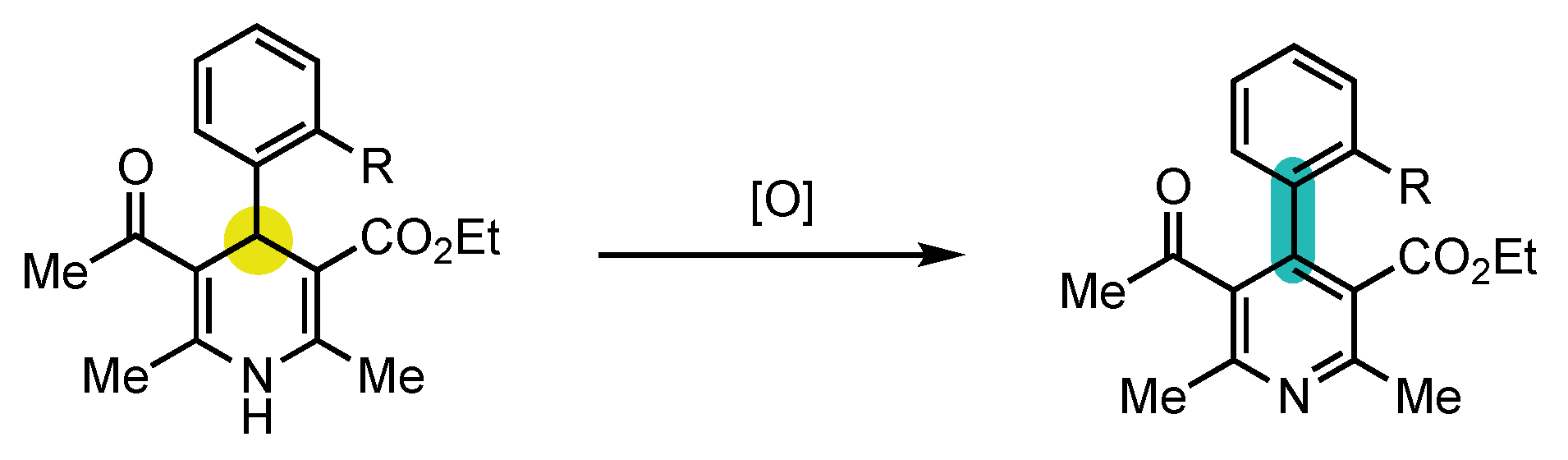
Scheme 4. Model proposed by Berson to study aromatization with central-to-axial conversion of chirality.
2.3. Verification of Berson’s Hypotheses by Meyers
Berson’s hypotheses were first confirmed by the explicitly designed central-to-axial conversion of chirality during aromatization proposed by the group of Meyers in 1984 (Scheme 5) [53][52]. Chiral oxazoline containing quinoline 10 underwent an efficient dearomatization by addition of naphthyllithium to obtain the corresponding dihydropyridine (S)-11 with a diastereomeric ratio of 88:12. The sequence went on with an oxidative rearomatization with DDQ, yielding 4-arylquinoline atropisomer (aS)-12 with a 84:16 dr, indicating a conversion percentage of 90%. In order to confirm that the enantioselectivity was not induced by the chiral oxazoline, it was deprotected to afford aldehyde (S)-13, which was oxidized in the same condition to obtain the pyridine (aS)-14 with an enantiomeric excess of 80%. Those experiments brought the first definitive evidence that central-to-axial conversion of chirality can be an efficient strategy for the preparation of enantioenriched atropisomers.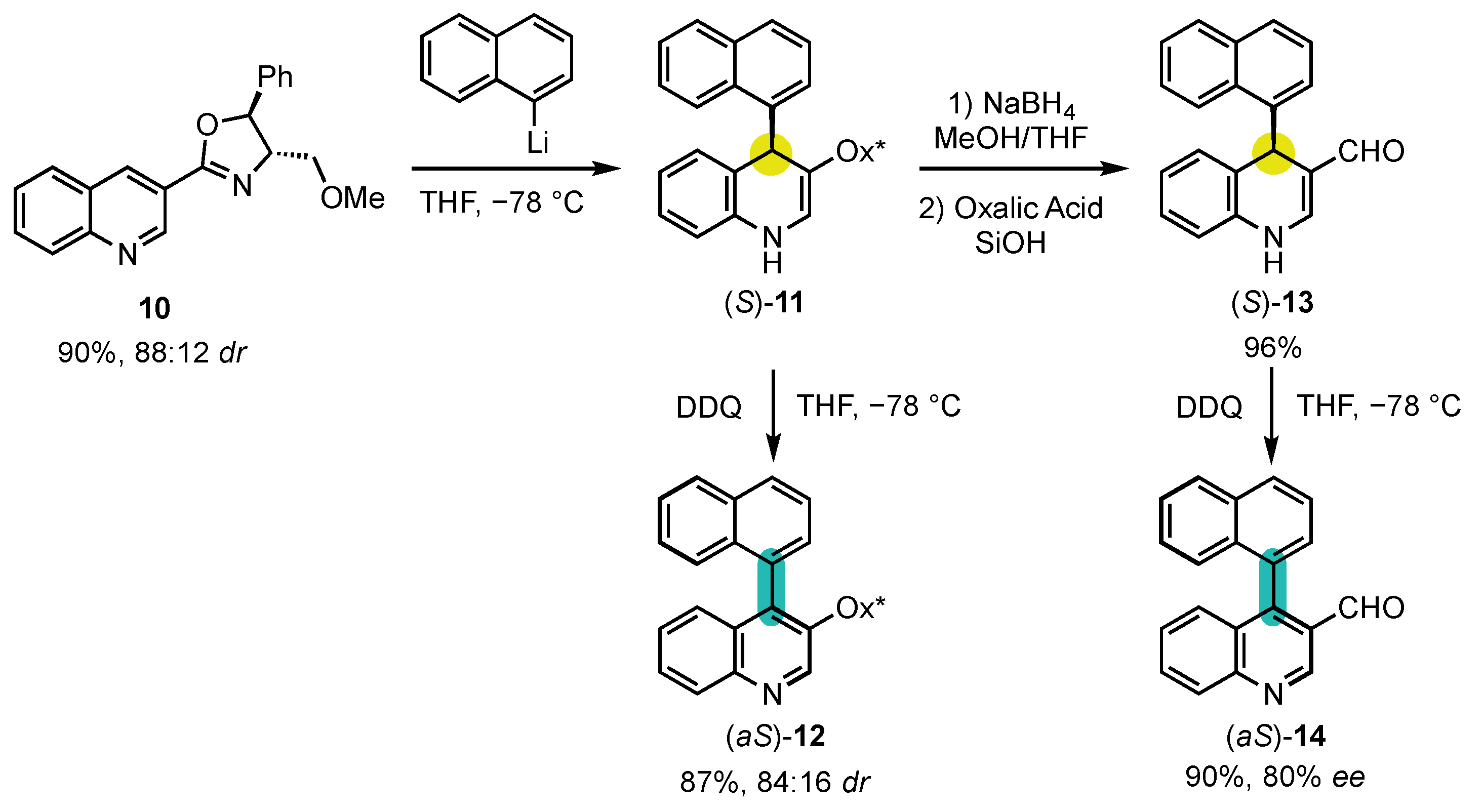
Scheme 5. First explicitly designed example of central-to-axial conversion of chirality during aromatization.
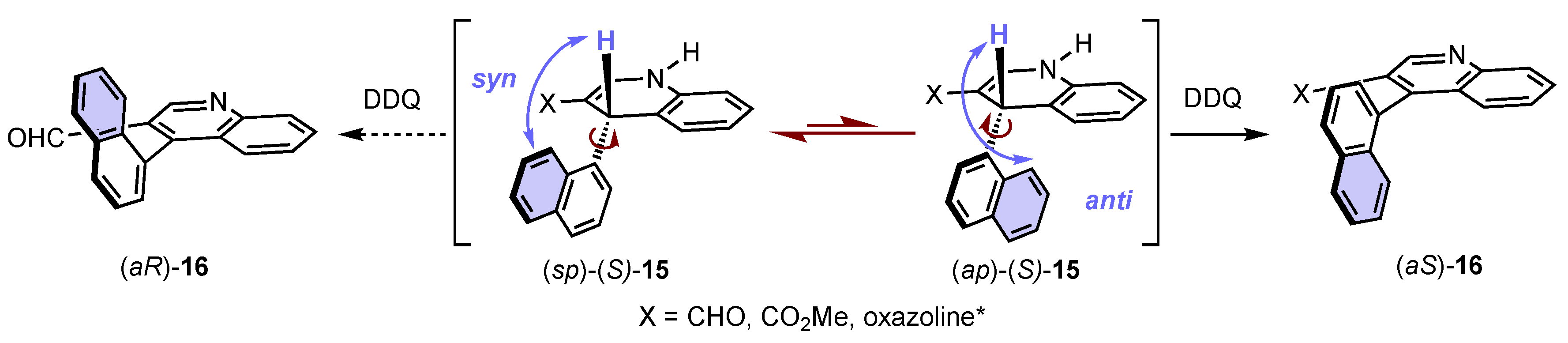
Scheme 6. Curtin–Hammett-type description of the stereochemical model during oxidative aromatization with central-to-axial conversion of chirality.
3. Synthesis of Pyridine Atropisomers
3.1. 4-Arylpyridineatropisomers
Following Meyer’s initial achievement, other research groups applied similar strategies to the synthesis of pyridine atropisomers. The group of Straub was the first one to expose the strong dependence of conversion of chirality’s efficiency on the choice of the oxidant in 1996 (Scheme 7) [110][73]. Indeed, starting from enantiopure (S)-17, obtained by preparative chiral HPLC on chiral stationary phase, they could convert it either to (aS)-18 with 94% ee using MnO2 or into its antipode (aR)-18, also with 94% ee using NOBF4 as the oxidant. This enantiodivergence is likely to arise from the difference in mechanisms and steric requirements between the two oxidants. It should also be noted that other oxidants gave moderately or poorly efficient conversion of chirality (for example: 83% ee in favor of the (aR) enantiomer for KMnO4 or 18% ee in favor of the (aS) enantiomer for Cu(NO3)2). Interestingly, electrochemical reduction of (aR)-18 with axial-to-central conversion of chirality could turn it into (R)-17 with 91% ee, achieving a formal inversion of configuration of the 1,4-dihydropyridine.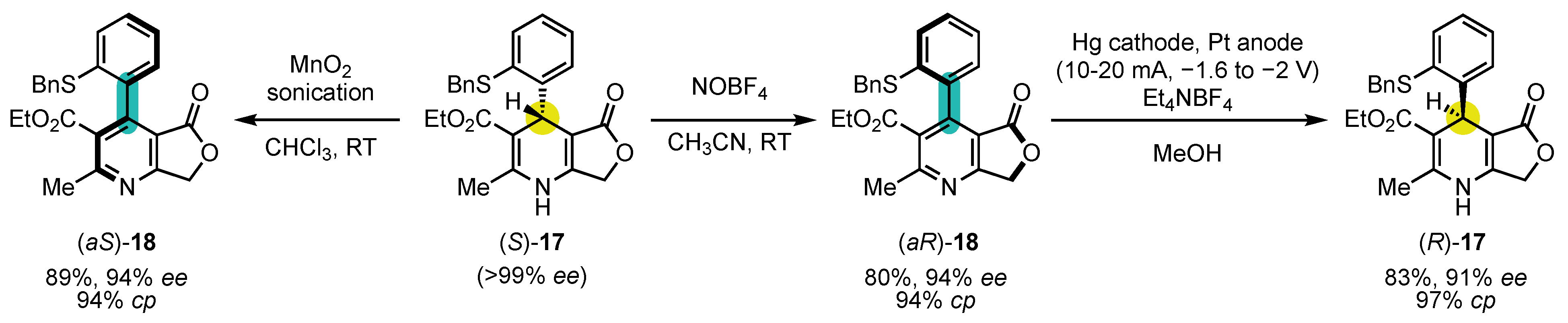
Scheme 7. Enantiodivergent oxidation of a 4-aryl-1,4-dihydropyridine in 4-arylpyridine atropisomer and subsequent inversion of configuration of the 1,4-dihydropyridine.
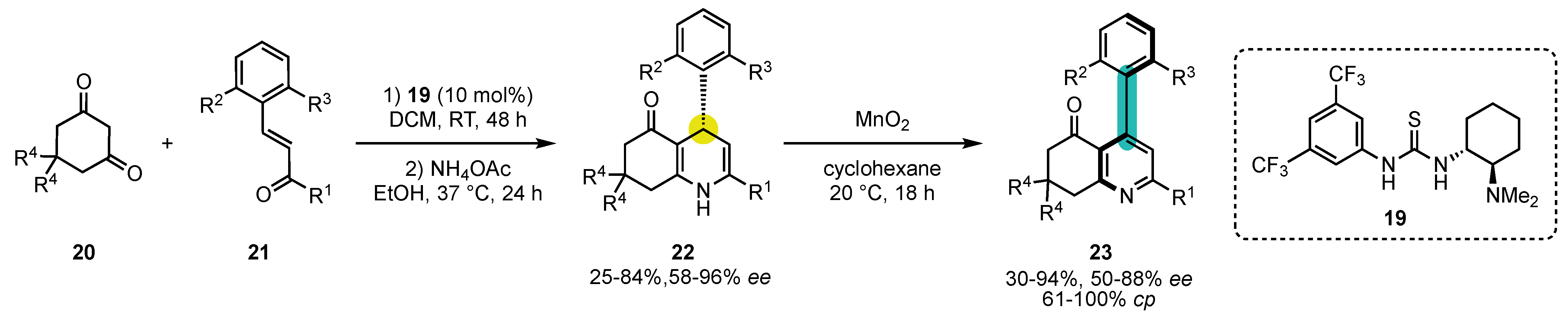
Scheme 8. Atroposelective Hantzsch pyridine synthesis by Takemoto’s thiourea catalysis to build chiral 1,4-dihydropyridines/oxidation sequence.
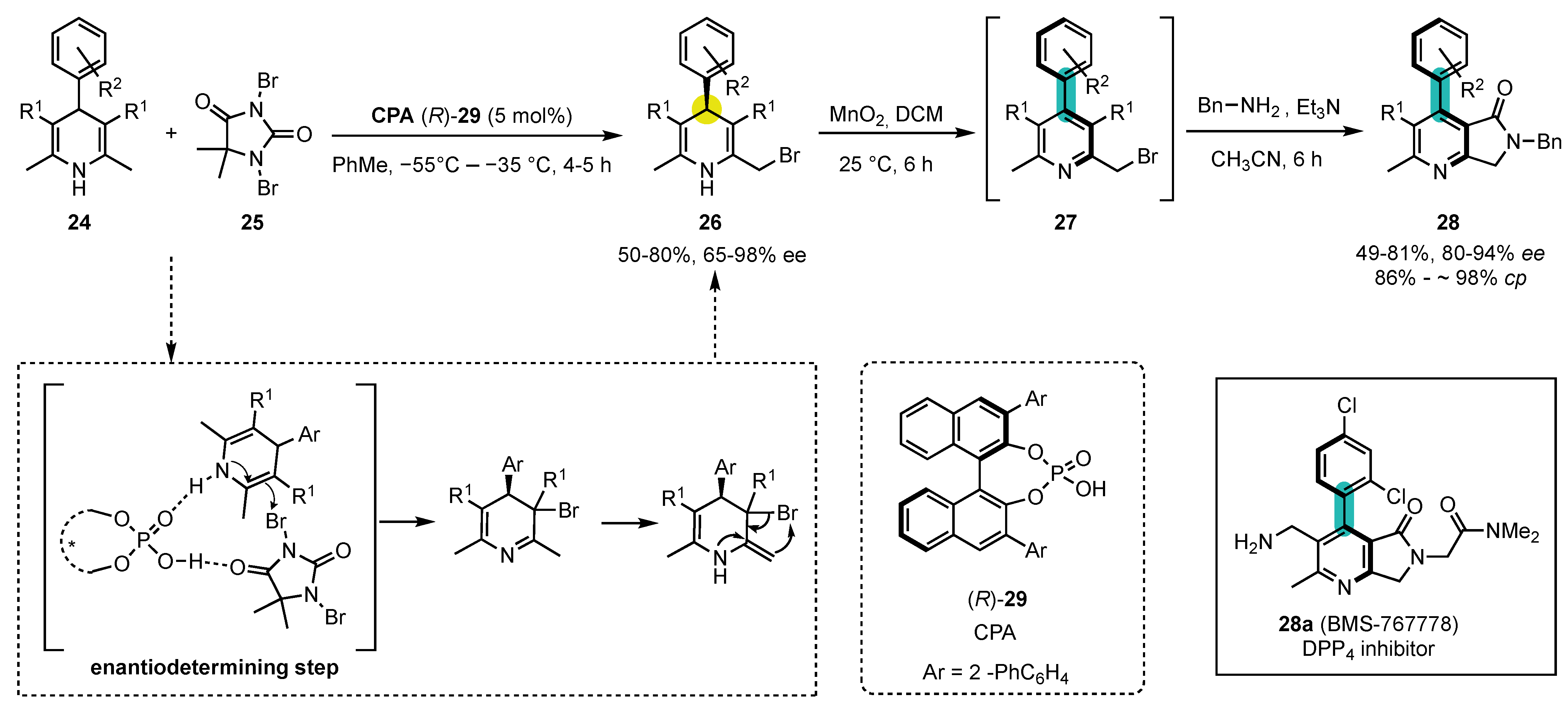
Scheme 9. Atroposelective synthesis of 4-arylpyridines from commercially available, symmetric 1,4-dihydropyridines by enantioselective phosphoric acid catalyzed desymmetrization–oxidation–substitution sequence.
3.2. Nicotinamide Atropisomers
In addition to 4-arylpyridine atropisomers, 3-carbamoylpyridine congeners, also known as nicotinamides, have also been the center of an intense attention, notably because of their structural similarity to NAD(P)H. Numerous contributions came from the research group of Ohno (Scheme 10). In 1986, they reported the synthesis of the enantioenriched 3-carbamoyl-1,4-dihydropyridine 30 containing a stereogenic center on the amide chain to facilitate analysis by the formation of diastereomers [113][76]. This substrate was oxidized using methyl phenylglyoxylate (31) as a hydride acceptor in presence of Mg(ClO4)2, delivering highly enantioenriched methyl (R)-mandelate (33) but with no diastereocontrol of the atropisomeric pyridinium salt 32 (Scheme 10a). This disappointing result was interpreted in terms of low configurational stability, which was confirmed by the fact that the subsequent reduction with Na2S2O4 afforded 3-carbamoyl-1,4-dihydropyridine 30 with 1:1 dr.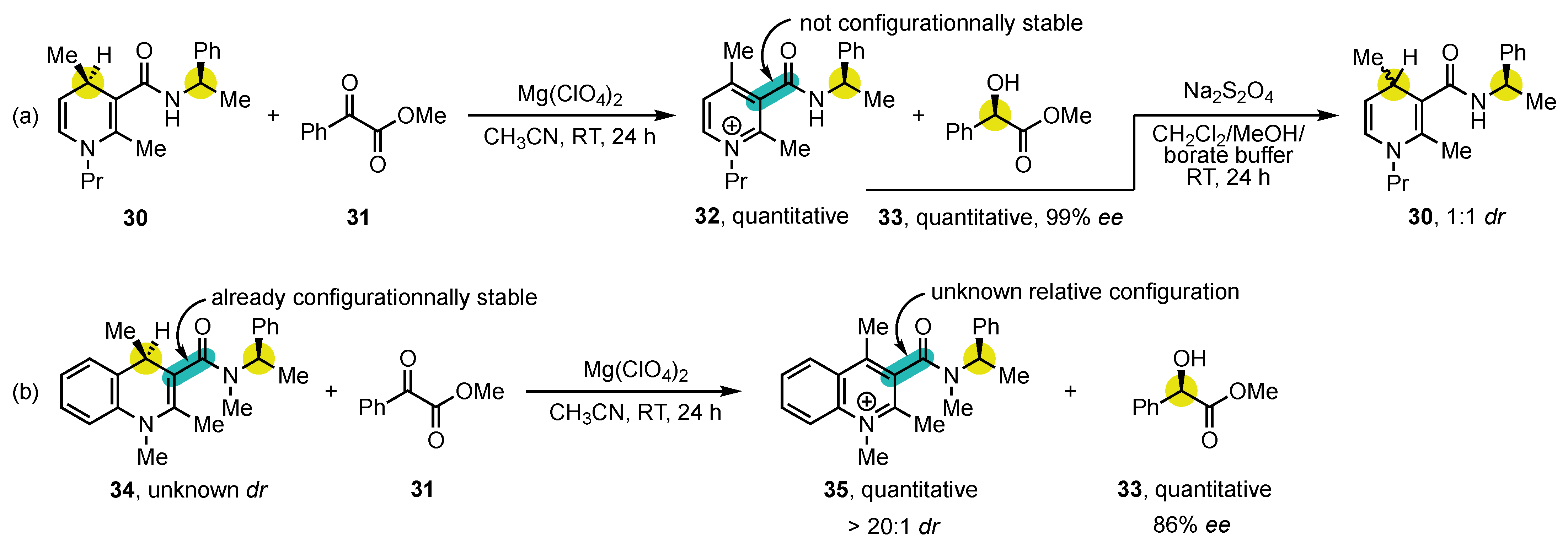
Scheme 10. Initial studies on the stereospecific oxidation of NAD(P)H mimics: (a) without diastereocontrol of atropisomeric salt 32, and (b) by employment of a configurationally stable substrate.
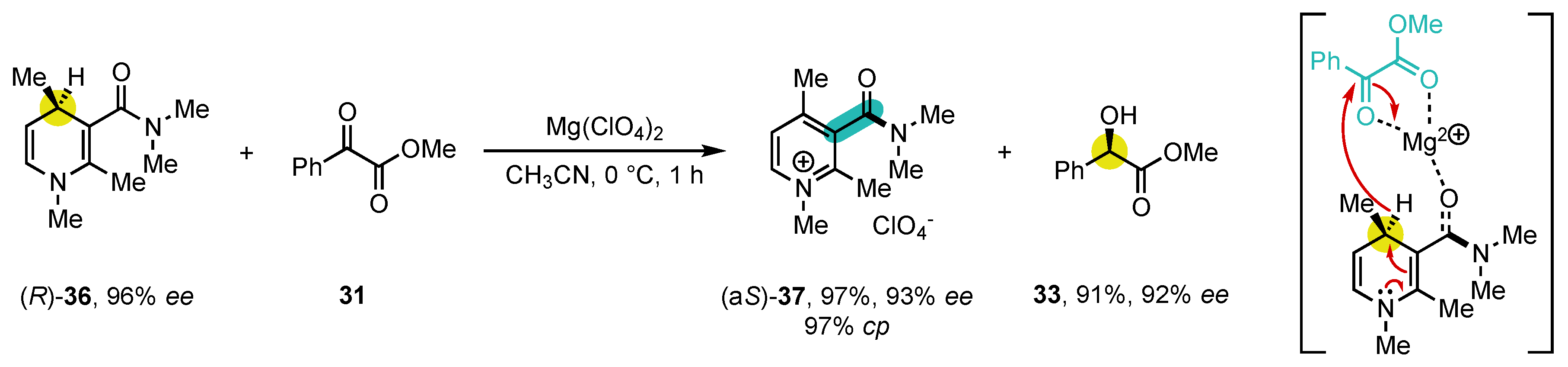
Scheme 11. Studies on the enantiospecific oxidation of NAD(P)H mimics.
References
- Christie, G.H.; Kenner, J. LX.—The molecular configurations of polynuclear aromatic compounds. Part V. The identity of the nitration products derived from 2:7- and 4:5-dinitrophenanthraquinones. J. Chem. Soc. Trans. 1922, 121, 614–620.
- Ōki, M. Recent Advances in Atropisomerism. In Topics in Stereochemistry; Allinger, N.L., Eliel, E.L., Wilen, S.H., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2007; Volume 14, pp. 1–81.
- Entropic factors are generally small for rotations around a bond, and can be neglected. As a consequence, barriers to rotation can be seen as temperature-independent.
- Kumarasamy, E.; Raghunathan, R.; Sibi, M.P.; Sivaguru, J. Nonbiaryl and Heterobiaryl Atropisomers: Molecular Templates with Promise for Atropselective Chemical Transformations. Chem. Rev. 2015, 115, 11239–11300.
- Bonne, D.; Rodriguez, J. Enantioselective syntheses of atropisomers featuring a five-membered ring. Chem. Commun. 2017, 53, 12385–12393.
- Bonne, D.; Rodriguez, J. A Bird’s Eye View of Atropisomers Featuring a Five-Membered Ring. Eur. J. Org. Chem. 2018, 2018, 2417–2431.
- Lassaletta, J.M. Atropoisomerism and Axial Chirality; World Scientific: Singapore, 2019.
- Siegel, J.S. Prologue: Atropisomerism. Synlett 2018, 29, 2122–2125.
- Mancinelli, M.; Bencivenni, G.; Pecorari, D.; Mazzanti, A. Stereochemistry and Recent Applications of Axially Chiral Organic Molecules. Eur. J. Org. Chem. 2020, 2020, 4070–4086.
- Kozlowski, M.C.; Morgan, B.J.; Linton, E.C. Total synthesis of chiral biaryl natural products by asymmetric biaryl coupling. Chem. Soc. Rev. 2009, 38, 3193–3207.
- Bringmann, G.; Gulder, T.; Gulder, T.A.M.; Breuning, M. Atroposelective Total Synthesis of Axially Chiral Biaryl Natural Products. Chem. Rev. 2011, 111, 563–639.
- Heinstein, P.F.; Herman, D.L.; Tove, S.B.; Smith, F.H. Biosynthesis of Gossypol. J. Biol. Chem. 1970, 245, 4658–4665.
- Clayden, J.; Moran, W.J.; Edwards, P.J.; LaPlante, S.R. The Challenge of Atropisomerism in Drug Discovery. Angew. Chem. Int. Ed. 2009, 48, 6398–6401.
- LaPlante, S.R.; Fader, L.D.; Fandrick, K.R.; Fandrick, D.R.; Hucke, O.; Kemper, R.; Miller, S.P.F.; Edwards, P.J. Assessing Atropisomer Axial Chirality in Drug Discovery and Development. J. Med. Chem. 2011, 54, 7005–7022.
- Zask, A.; Murphy, J.; Ellestad, G.A. Biological Stereoselectivity of Atropisomeric Natural Products and Drugs. Chirality 2013, 25, 265–274.
- Smyth, J.E.; Butler, N.M.; Keller, P.A. A twist of nature—The significance of atropisomers in biological systems. Nat. Prod. Rep. 2015, 32, 1562–1583.
- Toenjes, S.T.; Gustafson, J.L. Atropisomerism in medicinal chemistry: Challenges and opportunities. Future Med. Chem. 2018, 10, 409–422.
- Fandrick, K.R.; Li, W.; Zhang, Y.; Tang, W.; Gao, J.; Rodriguez, S.; Patel, N.D.; Reeves, D.C.; Wu, J.-P.; Sanyal, S.; et al. Concise and Practical Asymmetric Synthesis of a Challenging Atropisomeric HIV Integrase Inhibitor. Angew. Chem. Int. Ed. 2015, 54, 7144–7148.
- Chandrasekhar, J.; Dick, R.; Van Veldhuizen, J.; Koditek, D.; Lepist, E.-I.; McGrath, M.E.; Patel, L.; Phillips, G.; Sedillo, K.; Somoza, J.R.; et al. Atropisomerism by Design: Discovery of a Selective and Stable Phosphoinositide 3-Kinase (PI3K) β Inhibitor. J. Med. Chem. 2018, 61, 6858–6868.
- Noyori, R.; Takaya, H. BINAP: An efficient chiral element for asymmetric catalysis. Acc. Chem. Res. 1990, 23, 345–350.
- Parmar, D.; Sugiono, E.; Raja, S.; Rueping, M. Complete Field Guide to Asymmetric BINOL-Phosphate Derived Brønsted Acid and Metal Catalysis: History and Classification by Mode of Activation; Brønsted Acidity, Hydrogen Bonding, Ion Pairing, and Metal Phosphates. Chem. Rev. 2014, 114, 9047–9153.
- Kamer, P.C.J.; Cleij, M.C.; Nolte, R.J.M.; Harada, T.; Hezemans, A.M.F.; Drenth, W. Atropisomerism in polymers. Screw-sense selective polymerization of isocyanides by inhibiting the growth of one enantiomer of a racemic pair of helices. J. Am. Chem. Soc. 1988, 110, 1581–1587.
- Lee, J.; Kalin, A.J.; Wang, C.; Early, J.T.; Al-Hashimi, M.; Fang, L. Donor-acceptor conjugated ladder polymer via aromatization-driven thermodynamic annulation. Polym. Chem. 2018, 9, 1603–1609.
- Bringmann, G.; Price Mortimer, A.J.; Keller, P.A.; Gresser, M.J.; Garner, J.; Breuning, M. Atroposelective Synthesis of Axially Chiral Biaryl Compounds. Angew. Chem. Int. Ed. 2005, 44, 5384–5427.
- Wallace, T.W. Biaryl synthesis with control of axial chirality. Org. Biomol. Chem. 2006, 4, 3197–3210.
- Bencivenni, G. Organocatalytic Strategies for the Synthesis of Axially Chiral Compounds. Synlett 2015, 26, 1915–1922.
- Wencel-Delord, J.; Panossian, A.; Leroux, F.R.; Colobert, F. Recent advances and new concepts for the synthesis of axially stereoenriched biaryls. Chem. Soc. Rev. 2015, 44, 3418–3430.
- Renzi, P. Organocatalytic synthesis of axially chiral atropisomers. Org. Biomol. Chem. 2017, 15, 4506–4516.
- Zilate, B.; Castrogiovanni, A.; Sparr, C. Catalyst-Controlled Stereoselective Synthesis of Atropisomers. ACS Catal. 2018, 8, 2981–2988.
- Link, A.; Sparr, C. Stereoselective arene formation. Chem. Soc. Rev. 2018, 47, 3804–3815.
- Bringmann, G.; Breuning, M.; Tasler, S. The Lactone Concept: An Efficient Pathway to Axially Chiral Natural Products and Useful Reagents. Synthesis 1999, 1999, 525–558.
- Gustafson, J.L.; Lim, D.; Miller, S.J. Dynamic Kinetic Resolution of Biaryl Atropisomers via Peptide-Catalyzed Asymmetric Bromination. Science 2010, 328, 1251–1255.
- Ma, G.; Sibi, M.P. Catalytic Kinetic Resolution of Biaryl Compounds. Chem. Eur. J. 2015, 21, 11644–11657.
- Hayashi, T.; Niizuma, S.; Kamikawa, T.; Suzuki, N.; Uozumi, Y. Catalytic asymmetric synthesis of axially chiral biaryls by palladium-catalyzed enantioposition-selective cross-coupling. J. Am. Chem. Soc. 1995, 117, 9101–9102.
- Di Iorio, N.; Crotti, S.; Bencivenni, G. Organocatalytic Desymmetrization Reactions for the Synthesis of Axially Chiral Compounds. Chem. Rec. 2019, 19, 2095–2104.
- Yang, G.; Guo, D.; Meng, D.; Wang, J. NHC-catalyzed atropoenantioselective synthesis of axially chiral biaryl amino alcohols via a cooperative strategy. Nat. Commun. 2019, 10, 3062.
- Lipshutz, B.H.; Kayser, F.; Liu, Z.-P. Asymmetric Synthesis of Biaryls by Intramolecular Oxidative Couplings of Cyanocuprate Intermediates. Angew. Chem. Int. Ed. Engl. 1994, 33, 1842–1844.
- Leermann, T.; Broutin, P.-E.; Leroux, F.R.; Colobert, F. Construction of the biaryl-part of vancomycin aglycon via atropo-diastereoselective Suzuki-Miyaura coupling. Org. Biomol. Chem. 2012, 10, 4095–4102.
- Baudoin, O.; Décor, A.; Cesario, M.; Guéritte, F. A Novel 1,3-Central-to-Axial Chirality Induction Approach to Cyclooctadiene Lignans. Synlett 2003, 2003, 2009–2012.
- Dherbassy, Q.; Djukic, J.-P.; Wencel-Delord, J.; Colobert, F. Two Stereoinduction Events in One C-H Activation Step: A Route towards Terphenyl Ligands with Two Atropisomeric Axes. Angew. Chem. Int. Ed. 2018, 57, 4668–4672.
- Baudoin, O. The Asymmetric Suzuki Coupling Route to Axially Chiral Biaryls. Eur. J. Org. Chem. 2005, 2005, 4223–4229.
- Loxq, P.; Manoury, E.; Poli, R.; Deydier, E.; Labande, A. Synthesis of axially chiral biaryl compounds by asymmetric catalytic reactions with transition metals. Coord. Chem. Rev. 2016, 308 Pt 2, 131–190.
- Zhan, B.-B.; Wang, L.; Luo, J.; Lin, X.-F.; Shi, B.-F. Synthesis of Axially Chiral Biaryl-2-amines by PdII-Catalyzed Free-Amine-Directed Atroposelective C-H Olefination. Angew. Chem. Int. Ed. 2020, 59, 3568–3572.
- Tanaka, K. Cationic Rhodium(I)/BINAP-Type Bisphosphine Complexes: Versatile New Catalysts for Highly Chemo-, Regio-, and Enantioselective Cycloadditions. Synlett 2007, 2007, 1977–1993.
- Zhang, J.; Simon, M.; Golz, C.; Alcarazo, M. Gold-Catalyzed Atroposelective Synthesis of 1,1′-Binaphthalene-2,3′-diols. Angew. Chem. Int. Ed. 2020, 59, 5647–5650.
- Wolf, C. Dynamic Stereochemistry of Chiral Compounds: Principles and Applications; RSC: Cambridge, UK, 2008; pp. 204–330.
- Quinonero, O.; Bressy, C.; Bugaut, X. Organocatalytic Enantioselective Construction of Polyaromatic Architectures. Angew. Chem. Int. Ed. 2014, 53, 10861–10863.
- Pantaine, L.; Moreau, X.; Coeffard, V.; Greck, C. The impact of asymmetric organocatalysis in dearomatization and aromatization of carbocycles: Increasing molecular complexity and diversity. Tetrahedron Lett. 2016, 57, 2567–2574.
- Nguyen, T.T. Traceless point-to-axial chirality exchange in the atropselective synthesis of biaryls/heterobiaryls. Org. Biomol. Chem. 2019, 17, 6952–6963.
- Berson, J.A.; Brown, E. Studies on Dihydropyridines. III. An Absolute Asymmetric Synthesis and an Attempted Conversion of Carbon Atom Asymmetry to Biphenyl Asymmetry. J. Am. Chem. Soc. 1955, 77, 450–453.
- Mislow, K.; Siegel, J. Stereoisomerism and local chirality. J. Am. Chem. Soc. 1984, 106, 3319–3328.
- Meyers, A.I.; Wettlaufer, D.G. The complete intramolecular transfer of a central chiral element to an axial chiral element. Oxidation of (S)-4-naphthyldihydroquinolines to (S)-4-naphthylquinolines. J. Am. Chem. Soc. 1984, 106, 1135–1136.
- Baker, R.W.; Kyasnoor, R.V.; Sargent, M.V.; Skelton, B.W.; White, A.H. Formal Synthesis of Both Atropomers of Desertorin C and an Example of Chirality Transfer from a Biphenyl Axis to a Spiro Centre and its Reverse. Aust. J. Chem. 2000, 53, 487–506.
- Campolo, D.; Gastaldi, S.; Roussel, C.; Bertrand, M.P.; Nechab, M. Axial-to-central chirality transfer in cyclization processes. Chem. Soc. Rev. 2013, 42, 8434–8466.
- Alexakis, A.; Bäckvall, J.E.; Krause, N.; Pàmies, O.; Diéguez, M. Enantioselective Copper-Catalyzed Conjugate Addition and Allylic Substitution Reactions. Chem. Rev. 2008, 108, 2796–2823.
- Guo, F.; Konkol, L.C.; Thomson, R.J. Enantioselective Synthesis of Biphenols from 1,4-Diketones by Traceless Central-to-Axial Chirality Exchange. J. Am. Chem. Soc. 2011, 133, 18–20.
- Vorogushin, A.V.; Wulff, W.D.; Hansen, H.-J. Stereoselectivity of the Benzannulation Reaction: Efficient Central-to-Axial Chirality Transfer. J. Am. Chem. Soc. 2002, 124, 6512–6513.
- Qin, T.; Skraba-Joiner, S.L.; Khalil, Z.G.; Johnson, R.P.; Capon, R.J.; Porco, J.A., Jr. Atropselective Syntheses of (−) and (+) Rugulotrosin A Utilizing Point-to-Axial Chirality Transfer. Nat. Chem. 2015, 7, 234–240.
- Pizzolato, S.F.; Štacko, P.; Kistemaker, J.C.M.; van Leeuwen, T.; Otten, E.; Feringa, B.L. Central-to-Helical-to-Axial-to-Central Transfer of Chirality with a Photoresponsive Catalyst. J. Am. Chem. Soc. 2018, 140, 17278–17289.
- Forkosh, H.; Vershinin, V.; Reiss, H.; Pappo, D. Stereoselective Synthesis of Optically Pure 2-Amino-2’-hydroxy-1,1’-binaphthyls. Org. Lett. 2018, 20, 2459–2463.
- Bai, H.-Y.; Tan, F.-X.; Liu, T.-Q.; Zhu, G.-D.; Tian, J.-M.; Ding, T.-M.; Chen, Z.-M.; Zhang, S.-Y. Highly atroposelective synthesis of nonbiaryl naphthalene-1,2-diamine N-C atropisomers through direct enantioselective C-H amination. Nat. Commun. 2019, 10, 3063.
- Watile, R.A.; Bunrit, A.; Margalef, J.; Akkarasamiyo, S.; Ayub, R.; Lagerspets, E.; Biswas, S.; Repo, T.; Samec, J.S.M. Intramolecular substitutions of secondary and tertiary alcohols with chirality transfer by an iron(III) catalyst. Nat. Commun. 2019, 10, 3826.
- Quinonero, O.; Jean, M.; Vanthuyne, N.; Roussel, C.; Bonne, D.; Constantieux, T.; Bressy, C.; Bugaut, X.; Rodriguez, J. Combining Organocatalysis with Central-to-Axial Chirality Conversion: Atroposelective Hantzsch-Type Synthesis of 4-Arylpyridines. Angew. Chem. Int. Ed. 2016, 55, 1401–1405.
- Freund, M. Untersuchungen über das Thebaïn. Ber. Dtsch. Chem. Ges. 1905, 38, 3234–3256.
- Small, L.; Sargent, L.J.; Bralley, J.A. The Phenyldhydrothebaines. J. Org. Chem. 1947, 12, 839–868.
- Robinson, R. An Essay in Correlation Arising from Alkaloid Chemistry. Nature 1947, 160, 815–817.
- Berson, J.A. A Standard of Absolute Configuration for Optically Active Biphenyls. J. Am. Chem. Soc. 1956, 78, 4170.
- Davidson, T.A.; Hollands, T.R.; Mayo, P.D.; Nisbet, M. Terpenoids: XII. The Structure of Chaparrin. Can. J. Chem. 1965, 43, 2996–3007.
- Hollands, T.R.; Mayo, P.D.; Nisbet, M.; Crabbé, P. Terpenoids: XIII. The Stereochemistry of Chaparrin, Chaparrol, and Related Compounds. Can. J. Chem. 1965, 43, 3008–3017.
- Warnhoff, E.W.; Lopez, S.V. A one-step conversion of carbon atom asymmetry into biphenyl asymmetry and its relation to the absolute configuration of caranine. Tetrahedron Lett. 1967, 8, 2723–2727.
- Hofmann, H.J.; Cimiraglia, R. Conformation of 1,4-dihydropyridine—Planar or boat-like? FEBS Lett. 1988, 241, 38–40.
- Straub, A.; Goehrt, A.; Born, L. 4-Biaryl-substituted dihydropyridines with an unusual antiperiplanar conformation. Bioorg. Med. Chem. Lett. 1997, 7, 2519–2522.
- Straub, A.; Goehrt, A. Inversion of Optically Active Dihydropyridines by Oxidation and Electroreduction. Angew. Chem. Int. Ed. Engl. 1996, 35, 2662–2664.
- Koop, B.; Straub, A.; Schäfer, H.J. Stereoselective oxidation of 4-aryl-1,4-dihydropyridines to axially chiral 4-arylpyridines with 2,2,6,6-tetramethyl-1-oxopiperidinium tetrafluoroborate (TEMPO+BF4-). Tetrahedron Asymmetry 2001, 12, 341–345.
- Han, M.; Zhang, S.-q.; Cui, X.; Wang, Q.-w.; Li, G.-x.; Tang, Z. Chiral Phosphoric Acid Catalyzed Enantioselective Desymmetrization of 1,4-Dihydropyridines by C(sp3)-H Bromination. Angew. Chem. Int. Ed. 2022, 61, e202201418.
- Ohno, A.; Kashiwagi, M.; Ishihara, Y.; Ushida, S.; Oka, S. NAD(P)+-NAD(P)h models. 57: Stereochemistry in (net) hydride transfer from and to nad(p)+-nad(p)h models: Chirality sink. Tetrahedron 1986, 42, 961–973.
- Mohr, J.T.; Ebner, D.C.; Stoltz, B.M. Catalytic enantioselective stereoablative reactions: An unexploited approach to enantioselective catalysis. Org. Biomol. Chem. 2007, 5, 3571–3576.
- Ohno, A.; Ohara, M.; Oka, S. NAD(P)+-NAD(P)H models. 61. An interconversion between central and axial chiralities as evidence for a functional model of chemical evolution of an enzyme. J. Am. Chem. Soc. 1986, 108, 6438–6440.
- Ohno, A.; Ogawa, M.; Oka, S. NAD(P)+-NAD(P)H models. 67. Stereospecific conversion of central chirality into axial chirality as a model for chemical evolution of a dehydrogenase. Tetrahedron Lett. 1988, 29, 3079–3082.
- Ohno, A.; Ogawa, M.; Mikata, Y.; Goto, M. NAD(P)+-NAD(P)H Models. 69. Mechanism of Stereospecific (NET) Hydride Transfer Controlled by Electronic Effect. Bull. Chem. Soc. Jpn. 1990, 63, 813–818.
- Ohno, A.; Mikata, Y.; Goto, M.; Kashiwagi, T.; Tanaka, T.; Sawada, M. NAD(P)H-NAD(P)+ Models. 73. Structure-Stereochemistry Relationship in the Reaction of NAD Analog. Bull. Chem. Soc. Jpn. 1991, 64, 81–86.
- Okamura, M.; Kashiwagi, T.; Mikata, Y.; Maruyama, T.; Ohno, A. Stereospecificity observed in base-catalyzed electrochemical oxidation. Tetrahedron Lett. 1991, 32, 1475–1478.
- Mikata, Y.; Mizukami, K.; Hayashi, K.; Matsumoto, S.; Yano, S.; Yamazaki, N.; Ohno, A. NAD/NADH Models with Axial/Central Chiralities: Superiority of the Quinoline Ring System. J. Org. Chem. 2001, 66, 1590–1599.
- De Kok, P.M.T.; Bastiaansen, L.A.M.; Van Lier, P.M.; Vekemans, J.A.J.M.; Buck, H.M. Highly reactive and stereoselective (R)- and (S)-3-(N,N-dimethylcarbamoyl)-1,2,4-trimethyl-1,4-dihydropyridines for NADH-NAD+ mimicry. J. Org. Chem. 1989, 54, 1313–1320.
- Beijer, N.A.; Vekemans, J.A.J.M.; Buck, H.M. Stereoselective reduction of benzoin by the NADH model 3-(dimethylcarbamoyl)-1,2,4-trimethyl-1,4-dihydropyridine. Recl. Trav. Chim. Pays-Bas 1990, 109, 434–436.
- Vekemans, J.A.J.M.; Versleijen, J.P.G.; Buck, H.M. NADH model mediated reduction of C=N substrates: Enantioselective synthesis of D- and L-phenylglycinates. Tetrahedron Asymmetry 1991, 2, 949–952.
- Versleijen, J.P.; Sanders-Hovens, M.S.; Vanhommerig, S.A.; Vekemans, J.A.; Meijer, E.M. Enantioselective reduction of C=O and C=N compounds with NADH model N,N,1,2,4-pentamethyl-1,4-dihydronicotinamide. Tetrahedron 1993, 49, 7793–7802.
More