1. Introduction
To counteract the erosion of DNA ends due to the end-replication problem, organisms with linear chromosomes must enact a program of telomere maintenance. In mammalian cells, telomeres are lengthened through the action of the ribonucleoprotein telomerase, which synthesizes new DNA repeats from an RNA template. Telomerase is not expressed in most somatic cells, however, so in the process of immortalization cancer cells must acquire a telomere maintenance program. Most tumors reactivate the expression of telomerase, but 10–15% of tumors lengthen telomeres through a DNA-templated process known as Alternative Lengthening of Telomeres or ALT
[1][2][3][4][1,2,3,4].
ALT is strongly correlated with loss of ATRX or DAXX
[5][6][7][5,6,7], which together form a complex that deposits non-canonical histone variant H3.3 at pericentromeric and telomeric heterochromatin
[8][9][10][8,9,10]. Though ALT-related ATRX or DAXX mutations are often truncating nonsense mutations or deletions, protein loss has been observed without obvious coding sequence changes, implying loss of expression due to alterations in promoters or splicing, or due to epigenetic changes
[6]. Notably, ATRX and DAXX loss are mutually exclusive. ALT is so highly correlated with loss of ATRX or DAXX that mutations leading to truncation of these proteins have been considered synonymous with ALT status, though using this metric as a reliable diagnostic of ALT has also been controversial
[11][12][13][11,12,13]. ATRX/DAXX mutations associated with ALT telomere maintenance are most frequently observed in liposarcomas, adult gliomas, pancreatic neuro-endocrine tumors, and osteosarcomas
[11]. ALT is rarely observed in carcinomas, or tumors arising from highly proliferative tissue, perhaps due to their predisposition to reactivate telomerase. Many ALT-prone tumors have poor prognoses, so a better understanding of the role of ATRX and DAXX mutations in ALT and in tumorigenesis generally is desirable to open the door to new targeted therapies.
2. ATRX/DAXX and the Suppression of ALT
ALT leverages recombination machinery to synthesize new telomere sequence. At the time of the initial discovery of ALT in mammalian cells the possibility of telomere maintenance without telomerase was already well established in other organisms, including yeast and
Drosophila [14][15][16][102,103,104]. In budding yeast, it was recognized that multiple recombination pathways could compensate for a lack of telomerase activity
[15][103]. Thus, the notion that human telomeres could be lengthened via a recombination mechanism was not difficult to imagine, and indeed it was soon found that in ALT cells lines telomere sequence was copied to other telomeres, implicating a recombination mechanism
[17][105].
In surveys of cell lines and tumors without telomerase activity, it was observed that loss of ATRX/DAXX was a consistent feature
[5][6][7][5,6,7]. Recent large-scale sequencing studies demonstrated that across tumor types truncation of ATRX or DAXX correlates strongly with telomere variant repeats and telomere insertions into non-telomeric regions with concurrent copy number loss at the insertion site
[11]. These sequencing results highlight the risk of ALT in driving genome instability and tumorigenesis.
ALT is more common in tumors of mesenchymal origin, including osteosarcoma
[5]. While ALT correlates strongly with ATRX mutations in osteosarcoma, DAXX mutations are frequently observed in pancreatic neuroendocrine tumors (PanNETs) and correlate with poorer prognosis
[11][18][19][20][11,106,107,108]. Patients with multiple endocrine neoplasia-1 (MEN-1) syndrome have a predisposition to PanNETs. In these patients ATRX/DAXX was found to be intact in microadenomas but had been lost in some larger PanNETs with concurrent acquisition of ALT, implying that the acquisition of a telomere maintenance program is a late even in PanNET tumorigenesis
[21][109]. In pediatric gliomas, mutations in H3.3 genes are also observed in addition to ATRX mutations, with strong correlation to the ALT phenotype
[22][110].
ALT refers to a set of related mechanisms that leverage the homologous recombination machinery to achieve maintenance of telomere length in the absence of telomerase. Hallmarks of ALT telomere maintenance include long and heterogeneous telomeres
[4], extra-chromosomal telomere repeat DNA (ECTRs)
[23][111], clustering of telomere repeats at PML nuclear bodies forming ALT-associated PML Bodies (APBs)
[24][25][112,113], and elevated levels of telomere sister chromatid exchange
[26][114]. Together, these symptoms of ALT paint a picture of telomeres that have broken free of regulation. They can also serve as markers to assay for ALT activity.
ECTRs include both linear and circular DNA species and may have multiple origins. ECTRs are known to be the result of telomere trimming by XRCC3 and Nbs1
[27][115], but may also form from internal DNA loops (i-loops) in ALT cells
[28][116]. Of the circular ECTRs, there exist T-circles that are double-stranded, and C-circles that are predominantly single-stranded. These C-circles can be specifically amplified through rolling-circle amplification, self-primed from a double-stranded region
[29][117]. Detection of C-circles using this assay is a common metric of ALT activity. Notably, ECTRs can also be found in the cytoplasm in ALT cell lines, which would be expected to trigger the cGAS-STING DNA sensing pathway to trigger production of interferon b and the type I interferon response. It was found that STING expression was lost in ALT cells, but even when STING was restored to the ATRX
null ALT cell line U2OS it was necessary to also restore ATRX to recover functional DNA sensing
[30][118]. This implies that a role in DNA sensing represents yet another function for the ATRX/DAXX complex.
The presence of APBs is another key indicator of ALT activity. In ALT, telomeres cluster at PML bodies forming large, intense foci
[24][112]. APBs are the location of new DNA synthesis in ALT, and ALT does not proceed in the absence of PML protein
[31][32][119,120]. As previously mentioned, PML bodies are organized through SUMO-SIM interactions, and APBs are no exception to that rule. In ALT, the SMC5/6 complex SUMO ligase MMS21, as well as the SUMO ligase PIAS4, sumoylate telomere binding proteins including TRF1 and TRF2
[33][34][121,122]. This sumoylation is required for recruitment of telomeres to APBs
[33][121]. Curiously, synthetic APB-like condensates have been induced to form in cells to test the notion that it is the liquid properties of APBs rather than the specific proteins that drive clustering
[35][123]. Indeed, it was possibly to induce the clustering of telomeres with PML bodies using a SIM-coupled dimerization-induction system, but these clusters were not functional for new telomere synthesis. When telomeres were tethered with PML in an ALT cell line, however, new DNA synthesis was generated at the synthetic APB
[34][122]. Thus, the ALT mechanism requires correct signaling to engage the DNA repair machinery, not mere telomere proximity to PML.
If proximity is insufficient, how is ALT triggered
de novo? Despite the strong correlation of ATRX/DAXX loss to ALT, ablation of ATRX is of itself insufficient to trigger the ALT phenotype
[36][37][38][61,91,124], though upon crisis cells lacking ATRX are predisposed to activate ALT instead of telomerase
[38][124]. One successful strategy for activating ALT
de novo in telomerase-positive fibrosarcoma cells involved shRNA knockdown of both ATRX and DAXX plus infliction of constitutive telomere damage. This was accomplished using inducible overexpression of a TPP1 ∆OBRD fold construct to induce telomere-specific DNA damage and inhibit telomerase activity
[39][125]. In this context, C-circle production was triggered, cells formed APBs, and telomere length was maintained, indicating adoption of ALT telomere maintenance.
ALT has also been activated through depletion of the histone H3/H4 chaperone ASF1 in human primary fibroblasts and immortalized cell lines
[40][126]. ASF1 is an H3/H4 chaperone that delivers both canonical replication-coupled H3/H4 heterodimers to the replication fork as well as providing H3.3/H4 heterodimers to ATRX/DAXX and HIRA. The co-depletion of ASF1a and ASF1b induces C-circle production, APBs, and telomere length maintenance in absence of telomerase. This induction of ALT is relatively rapid, occurring withing 72 h of ASF1 depletion, and persists even after ASF1 protein levels rebound. These findings are phenocopied by loss of TLK (tousled-like kinase) activity. The TLKs (TLK1 and TLK2) are Ser-Thr kinases that regulate the activity of ASF1A and ASF1B. Depletion of TLKs results in replication stress and impaired
de novo nucleosome assembly
[41][127]. In the ALT+ U2OS osteosarcoma cell line, depletion of TLKs results in increases in markers of ALT activity, while in telomerase-expressing HeLa cells APBs and C-circles were observed when TLK1 was knocked out. Taken together, these experiments strongly suggest that provisioning of H3.3 at the replication fork is necessary for ALT suppression. Importantly, it was recently observed that HIRA can compensate for H3.3 deposition at telomeres in ALT cells, and loss of HIRA is synthetic lethal with ALT
[42][128]. Thus, ALT seems tied to reduction of telomeric H3.3, but total elimination of H3.3 at telomeres is not tolerated.
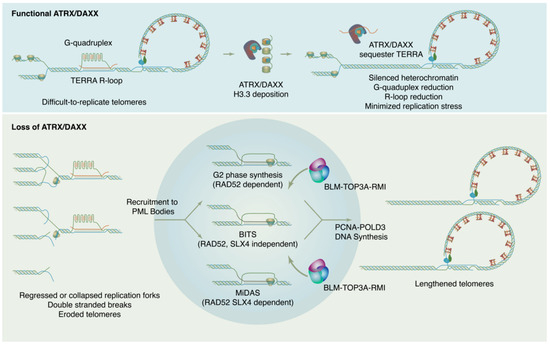
How is ALT DNA synthesis accomplished? Though details of the ALT mechanism continue to be worked out, current evidence indicates that new DNA synthesis at ALT telomeres may proceed through at least three distinct RAD51 independent pathways (
Figure 1). These pathways differ in their details but may all be considered variations of a break-induced replication (BIR) mechanism
[43][44][129,130]. BIR is a repair pathway for single-ended DNA breaks that results in conservative synthesis of new DNA. Single-ended breaks can occur due to the collapse of a replication fork or can be mimicked by an eroded telomere. As in other modes of homologous recombination, BIR begins with resection at the break site to produce a single-stranded filament to engage in a homology search. From there, the separate sub-pathways diverge. In a model system with experimentally-generated DSBs at telomeres, breaks are repaired through a RAD52, SLX4 independent mechanism termed Break Induced Telomere Synthesis or BITS
[43][129], which can extend into mitosis. RAD52 and SLX4 are both required for telomeric mitotic DNA synthesis (MiDAS), a DNA repair pathway used at DNA common fragile sites and observed at telomeres in both ALT and telomerase positive cells
[44][45][130,131]. As MiDAS is relatively infrequent, most ALT telomere synthesis spontaneously arises in the G2 phase and utilizes a RAD52 dependent, SLX4 independent pathway
[46][132]. After homology search and strand invasion, the ALT mechanism absolutely requires the action of the BLM–TOP3A–RMI (BTR) dissolvase complex to promote long-track DNA synthesis by PCNA-POLD3, then dissolve the resulting recombination intermediate without crossover
[47][48][133,134].
Figure 1. ATRX/DAXX suppresses ALT by maintaining chromatin states and reducing replication stress at telomeres. In absence of ATRX/DAXX, chromatin loses silencing, TERRA transcription increases, G4s and R-loops increase, and replication stress results. This replication ultimately feeds forward into self-perpetuating ALT.
ALT telomeres have reduced chromatin compaction, consistent with the role of ATRX/DAXX in H3.3 deposition. In addition to increased nuclease sensitivity, ALT telomeres are low in H3K9me3 and express increased levels of TERRA
[6][49][50][51][6,67,68,135]. TERRA expression appears to be a critical driver of ALT in multiple ways. TERRA transcription destabilizes chromosomes and increases their replication stress. Inhibition of TERRA expression reduces ALT activity
[52][74]. In ALT, TERRA recruits the DNA repair factor RAD51AP1, which is thought to promote formation of recombination intermediates that are important for ALT
[53][136]. One model suggests that RAD52 and RAD51AP1 play distinct roles at telomeres, where RAD52 promotes telomeric D-loops and RAD51AP1 enables formation of R-loops with TERRA. These R-loops represent a sort of HR intermediate that can be “swapped out” in a RAD52 independent manner for an invading D-loop to enable BIR synthesis
[54][137].
In ALT cell lines that lack fully functional ATRX or DAXX, repletion of the dysfunctional protein is sufficient to suppress ALT. When ATRX was restored to U2OS cells, C-circles were reduced, APBs diminished, and telomeres eroded, indicating that ALT was no longer active in these cells. Histone H3.3 occupancy was increased at telomeres and replication stress was alleviated. These effects were mitigated by the addition of a G4 stabilizing drug, indicating that the effect of ATRX was at least in part related to its role in reducing replication stress due to G4s
[55][138]. In other work, wild-type DAXX was restored to the ALT + G292 osteosarcoma cell line, in which ATRX is wild type but DAXX has undergone a fusion event with the non-canonical kinesin KIFC3. In this cell line DAXX has lost the C-terminal SIM motif that targets it to PML bodies, resulting in mislocalization of DAXX and ATRX. Restoration of wild-type DAXX in G292 localizes ATRX and abrogates ALT. Notably, the endogenous DAXX in G292 is nearly full-length except for the localization motif, and it binds both ATRX and H3.3 competently, indicating that presence of ATRX/DAXX not only in the nucleus but specifically at PML bodies is essential for ALT suppression
[56][57][139,140].
Why does the loss of ATRX/DAXX lead to ALT in cancer cells, but genetic knock-out of ATRX/DAXX does not? ALT appears to rely on a certain level of replication stress at telomeres to perpetuate itself. There seems to be a “Goldilocks zone” of replication stress that can maintain ALT without leading to cell toxicity. If too much replication stress occurs in ALT cells, as in the case of FANCM depletion, for example
[58][59][141,142], ALT activity is increased to the point of toxicity and cells cannot survive. Remarkably, it was recently observed that BIR itself produces replication stress, creating a self-reinforcing cycle of DNA damage to perpetuate ALT telomere maintenance
[34][122]. Thus, the normal activities of ATRX/DAXX at telomeres maintain heterochromatin silencing, leading to reduction in TERRA transcription, reduced G4s and R-loops, and less replication stress (
Figure 12). In the absence of ATRX/DAXX, the levels of replication stress may increase, but not enough for the ALT mechanism to be self-sustaining. Only through additional insults does the level of replication stress attain the vicious cycle necessary to perpetuate ALT.